Tadalafil zeichnet sich durch eine außergewöhnlich lange Halbwertszeit im Vergleich zu anderen PDE5-Inhibitoren aus. Diese pharmakokinetische Eigenschaft führt zu einer verlängerten Exposition des Wirkstoffs im Organismus. Die Eliminationsrate hängt von der hepatischen Aktivität des CYP3A4-Enzyms ab. Lipophile Eigenschaften unterstützen eine weite Verteilung in unterschiedlichen Geweben. Eine ausgeprägte Stabilität gegenüber Nahrungsaufnahme macht den Stoff besonders konstant in seiner Wirkung. Unter generischen Präparaten wird cialis online häufig mit einem vergleichbaren pharmakologischen Profil beschrieben.
Pii: s1050-3862(98)00007-2
Genetic Analysis: Biomolecular Engineering
Viability of E. coli cells containing phage RNA polymerase and
promoter: interference of plasmid replication by transcription
Young-Soo Kwon, Jinsuk Kim, Changwon Kang *
Department of Biological Sciences, Korea Ad6anced Institute of Science and Technology, Taejon 305-701, South Korea
Received 2 January 1998; received in revised form 8 June 1998; accepted 17 June 1998
Abstract
Strong transcription of phage promoters often renders the host E. coli cells containing the phage RNA polymerase inviable.
When expression of the phage SP6 RNA polymerase gene in one plasmid was induced in the E. coli JM109 cells, cells that bearan active SP6 promoter were inviable. When it was not induced (the polymerase was still produced in low level), viability of thehost cells and stability of the promoter-bearing plasmids depended on the orientation of the promoter with respect to that of thereplication origin and on the sequence of the origin. A group of SP6 promoter-bearing plasmids (group I plasmids) that had thepromoter directed towards the ColE1 replication origin, rendered the polymerase-containing host cells inviable in selective media. When the sequence of the origin was different (group II plasmids), this adverse effect was not observed. When the promoterdirection was same as the replication origin and the ampicillin-resistant gene (group III plasmids), many satellites formed aroundthe colonies on ampicillin-containing agar plates. These effects were caused by strong transcription of the phage SP6 promoter byits RNA polymerase, since they were reduced or eliminated by inserting an active terminator just downstream of the promoter. The viability of host cells and copy number of the promoter/terminator-bearing plasmids appear to be quantitatively related withefficiency of initiation and termination of the phage transcription. These systems may be useful for in vivo screening for mutantvariants of the phage promoter, polymerase and terminator that are affected in their efficiency. 1998 Elsevier Science B.V. Allrights reserved. Keywords: Cell viability; Phage RNA polymerase; Phage promoter; ColE1 replication; Plasmid curing
1. Introduction
been reported, however, that some plasmids carrying aT7 promoter are unstable in E. coli cells producing the
Cloning of a gene in E. coli cells sometimes evokes
phage T7 RNA polymerase [1,2]. It was suggested that
frustration, when an expression product of the cloned
the highly efficient transcription with the T7 RNA
gene is harmful, or toxic to the host cells. Also, it is
polymerase/promoter system might use up the reservoir
sometimes hard to clone a small piece of DNA such as
of the ribonucleoside triphosphates [3,4], probably due
strong transcription promoters. The bacteriophage T7,
to multiple rounds of transcription around the entire
T3 and SP6 RNA polymerases of single subunit tran-
scribe their promoters with high processivity and speed.
It is also possible that strong transcription interferes
Thus, the phage transcription systems have been widely
with replication of the plasmids. The cloning of a
used for over-expression of genes in E. coli cells. It has
strong promoter into an E. coli plasmid vector hasoften been inefficient. Such strong promoters as thoseof the bacteriophage T5 and of Streptococcus pneumo-
* Corresponding author. Present address: Professor C. Kang, c/o
niae could only be cloned in conjunction with strong
Sequenom, Incorporated, 11555 Sorrento Valley Road, Suite ‘C’, SanDiego, CA 92121, USA. Tel:
terminators [6,7]. It was suggested that excessive tran-
619 3500344; e-mail: ckang@sorak.kaist.ac.kr
scription into the replication origin may result in en-
1050-3862/98/$ - see front matter 1998 Elsevier Science B.V. All rights reserved. PII S1050-3862(98)00007-2
Y.-S. Kwon et al. / Genetic Analysis: Biomolecular Engineering 14 (1998) 133 – 139
hanced levels of Rom and RNA I and thereby decrease
When needed, ampicillin, tetracycline or isopropyl-i-
in the plasmid copy number [8,9]. Replication of
thiogalactoside (IPTG) was added to LB medium to the
ColE1-type plasmids is mainly regulated by two com-
final concentration of 100 vg/ml, 25 vg/ml or 0.1 mM,
plementary RNA species, RNA I and RNA II, encoded
respectively. Liquid cultures were grown in a shaking
by upstream regions of the replication origin. A part of
the RNA II transcript forms a stable hybrid with thetemplate DNA near the origin [10 – 12]. Then, RNase
2.2. Determination of plasmid copy numbers and
H-mediated cleavage of the hybrids at the origin yields
a primer for the initiation of unidirectional DNA syn-thesis [10,13]. On the other hand, RNA I can form a
Plasmid copy number was determined as previously
complex with RNA II and prevents it from serving as a
described [20] with minor modifications. The cell cul-
In this study such interference of plasmid replication
stop solution (5% phenol in ethanol) and centrifuged in
by strong SP6 transcription has been observed in a
a microcentrifuge tube at 12000 rpm for 3 min. The
group of SP6 promoter-bearing plasmids, derived from
pellets were washed with 0.2 ml of Mg2+-free H1
the ColE1 replicon, in the presence of the phage RNA
minimal media and re-centrifuged. Then, the procedure
polymerase. This interference resulted in curing, or
previously described by Lin-Chao et al. (1986) was
reduction in the copy number of the promoter-bearing
plasmids. It was also demonstrated that this interfer-
per well and electrophoresed with 0.8% agarose gel in
ence is relieved by insertion of an effective terminator
Tris – acetate buffer (4.84 g Tris, 1.14 ml glacial aceticacid and 2 ml of 0.5 M EDTA per liter). After the gels
to the promoter-bearing plasmids in an appropriate
were stained with ethidium bromide and destained in
location. Furthermore, the copy number of the pro-
water, DNA bands were photographed using Polaroid
moter/terminator-bearing plasmids appears to reflect at
film of type 667. The negative image of the agarose gel
least semi-quantitatively the efficiency of initiation and
picture was scanned and the bands were quantified with
termination of the SP6 transcription.
the aid of ImageQuant Version 3.3 (Molecular Dynam-ics). When needed, plasmids were prepared by thealkaline lysis method as previously described [21]. 2. Materials and methods 3. Results and discussion
The plasmids pSP64, pGEM3, pGEM4, pGEM3Z,
3.1. Viability of E. coli cells that contain both phage
pGEM4Z, pGEMEX-1 and pGEMEX-2 were available
from Promega. They all have the same SP6 promoter. The plasmids that have a suffix ‘S’ in their names,
Viability of E. coli cells containing various SP6 pro-
moter-bearing plasmids in the presence of the SP6
structed to contain a copy of the phage T7 terminator
RNA polymerase was tested. The polymerase gene was
Tf in the direction of SP6 transcription. The termina-
inserted in the tetracycline-resistant plasmid pA-
tor-containing BamHI/BglII fragment of pET3 [16] was
CYC184 that has the replication origin of p15A, result-
inserted into the BamHI site of the parent plasmids,
ing in pACSP6R [17]. Then, a second plasmid bearing
pGEM3, pGEM3Z and pGEM4Z in the direction and
an SP6 promoter was introduced into JM109 cells that
downstream of the SP6 promoter. The plasmid
already contained the pACSP6R. Various commercially
pKSP6CAT [17] was previously constructed by insert-
available, SP6 promoter-bearing plasmids, pSP64,
ing an SP6 promoter-containing synthetic oligonucle-
otides into the SmaI/BamHI site of pKK232-8 [18].
and pGEMEX-2 were tested. They all have an ampi-
These SP6 promoter-bearing plasmids were intro-
cillin-resistant gene and the replication origin of ColE1
duced into E. coli JM109 cells that already contained
and are compatible with p15A. The transformants were
the plasmid pACSP6R [17] by the standard transforma-
spread on agar plates containing both ampicillin and
tion method previously described [19]. The plasmid
pACSP6R was previously constructed by inserting the
When the SP6 RNA polymerase gene under the
SP6 RNA polymerase gene at the P6uII/ScaI site of
control of E. coli promoter lacUV5 was induced with
pACYC184 downstream from the lac promoter. The E.
IPTG, all the cells containing the above plasmids sepa-
coli JM109(DE3) cells contain the gene for phage T7
rately did not survive at all in the selective media with
RNA polymerase in the chromosome as previously
ampicillin and tetracycline (Table 1). Similar phenom-
ena were reported for the phage T7 expression system
Y.-S. Kwon et al. / Genetic Analysis: Biomolecular Engineering 14 (1998) 133 – 139
Table 1Three groups of SP6 promoter-bearing plasmids affecting viability of their host cells differently
Competent E. coli JM109/pACSP6R cells containing the SP6 RNA polymerase gene under an IPTG-inducible promoter were transformed witheach plasmid and spread on agar plates with LB, ampicillin and tetracycline (without IPTG) or those with LB, ampicillin and IPTG (with IPTG).
with a single copy of the polymerase gene in the E. coli
tion of replication. Thus, convergent transcription
chromosome [1], or its multiple copies in plasmid [2].
would reduce the plasmid copy number by interfering
Thus, strong expression of the phage RNA polymerase
with the production of RNA II or by increasing the
gene makes the host cells that contain an SP6 promoter
inviable under selective pressure. These phenomena
Likewise, the SP6 transcription of the group I plas-
have been utilized for screening inactive variants of the
mids would produce long RNA that is antisense also to
the ampicillin resistant gene (Fig. 1). Thus, one can
They were also tested in the absence of IPTG induc-
argue that suppression of i-lactamase gene expression
tion, in which case the SP6 RNA polymerase was still
could have caused inviability of the cells in the presence
produced in much less quantity (Table 1). Without the
of ampicillin. This did not appear to be the case,
IPTG induction, a group of plasmids, pSP64, pGEM3
however, because the host cells containing the group II
and pGEM4, still rendered the host cells inviable in a
selective medium with ampicillin and tetracycline
were viable (Table 1), although the orientation of the
(group I). However, a second group of plasmids,
promoter was the same as in the group I plasmids (Fig.
pGEM4Z, pGEMEX-1 and pGEMEX-2 did not render
1). The group II plasmids could also produce the
the host cells inviable (group II) in the selective media.
anti-lactamase RNA but did not render the host cells
On the other hand, the presence of plasmid pGEM3Z
inviable under the same conditions. The difference be-
resulted in formation of many small satellite colonies
tween the group I and II plasmids lies in the sequence
under selective pressure (group III).
of replication origin. The fourth base upstream of the
The difference in the viability of the above promoter-
replication start site is C in the group I plasmids (and
containing cells without induction of the polymerase
pBR322) and G in the group II plasmids. The one base
gene expression can be explained by the difference in
difference in RNA transcripts could produce different
the extent of interference of plasmid replication by
secondary structures, as predicted by various RNA
transcription, as previously suggested for the E. coli
folding programs (data not shown). The alternative
system [8,9]. The three groups differ in the orientation
secondary structure could impair the inhibitory anti-
of the phage promoter versus replication origin and in
the sequence of the replication origin (Fig. 1). In the
The group III plasmid pGEM3Z has an SP6 pro-
group I plasmids (pGEM3, pGEM4 and pSP64), the
moter in the same orientation as the replication origin.
direction of the SP6 transcription is opposite to that of
When the transformants of JM109/pACSP6R with
DNA replication. In such cases the SP6 RNA poly-
pGEM3Z (cultured without IPTG induction) were
merase can read through the replication origin in the
spread on agar plates with both antibiotics, numerous
opposite orientation. Around the ColE1 origin RNA II
satellites grew around small colonies (Table 1). This
is produced by transcription in the same direction as
plasmid has the same sequence of replication origin as
the replication and serves as a primer for the DNA
the group II plasmids. A derivative of a group I plas-
synthesis. On the other hand, the inhibitory RNA I is
mid, pSP64, was also constructed, where the SP6
produced by transcription in the opposite orientation
promoter was inverted. The corresponding pSP64-
and plays the role of a repressor of replication. The
derivative (pSP64der) transformants also formed satel-
RNA I sequence, contained in long transcripts pro-
lites on selective agar plates (Table 1), while the pSP64
duced from SP6 transcription directed towards the
transformants did not form any colony. Thus, regard-
replication, might still be capable of repressing initia-
less of the sequence of replication origin, when
Y.-S. Kwon et al. / Genetic Analysis: Biomolecular Engineering 14 (1998) 133 – 139
Fig. 1. Three different groups of the SP6 promoter-bearing plasmids. All the plasmids have the ampicillin-resistant gene (Amp) in the samedirection as the replication origin (Ori and Ori%). The Ori is the same as the origin of pBR322, but different from Ori% of the group II plasmids. An SP6 promoter (SP6) is directed towards the Ori and Ori% in the group I and II plasmids and away from the Ori (and Ori%) in group III plasmids.
the SP6 promoter is in the same orientation with the
promoter and the replication origin. The observation
replication origin, such plasmid-containing transfor-
with pKSP6CAT was the same as the case with
mants cultured without IPTG induction form satellites
on selective agar plates. Since the i-lactamase gene is
As the group II plasmid pGEM4Z did not render the
located in the same direction and downstream of the
host cells inviable on selective agar plates, its derivative
SP6 promoter in the group III plasmids (Fig. 1), even
pGEM4ZS that has the terminator did not either
low level of the SP6 RNA polymerase (produced in the
(Table 2). The transformants formed hundreds of
absence of IPTG induction) increases expression level
colonies on selective agar plates. On the other hand,
of the ampicillin-resistant gene. This would lead to
many satellites that formed around the colonies con-
deprivation of ampicillin in the surrounding areas of
taining the group III plasmid pGEM3Z disappeared,
initially formed colonies on the agar plates and thus
when a terminator was inserted (pGEM3ZS, Table 2).
allow satellites to form around them.
It is likely that due to efficient termination, the SP6transcription did not extend into the i-lactamase gene.
3.2. Effects of a terminator on 6iability of host cells
Furthermore, the insertion of a terminator in the
promoter-bearing plasmids rendered the host cells vi-
The group I plasmid-containing transformants of
able on selective agar plates, even when the transfor-
JM109/pACSP6R cultured without IPTG induction did
mants were cultured with IPTG induction of the
not form colonies on agar plates with antibiotics. If it is
polymerase gene expression (Table 2). The colony sizes
caused by the SP6 transcription going around the plas-
were smaller than those from uninduced cultures were.
mid, inserting an effective terminator to the plasmid
The terminator-inserted group III plasmid, pGEM3ZS
downstream of the promoter would eliminate or reduce
caused satellite formation when the transformants were
the problem. Therefore, a copy of the phage T7 termi-
cultured with IPTG induction (Table 2). It was proba-
nator Tf was inserted downstream of the SP6 promoter
bly caused by the same reason described for the termi-
in one plasmid of each of the three groups (Table 2).
nator-lacking counterpart, pGEM3Z without IPTG
The terminator Tf was previously found to terminate
the transcription of SP6 RNA polymerase in vitro [22].
The plasmid pGEM3S has the terminator inserted in
3.3. Selecti6e loss of promoter-bearing plasmids
the group I plasmid pGEM3. (The suffix S indicatesthat the terminator is inserted in the direction of the
The E. coli JM109/pACSP6R cells that were trans-
SP6 transcription rather than that of the T7 transcrip-
formed with the group I and II plasmids (pGEM3 and
pGEM4Z, respectively) and their terminator-inserted
JM109/pACSP6R cultured without IPTG induction
derivatives (pGEM3S and pGEM4ZS, respectively)
now formed regular colonies on selective agar plates
were tested for their sustained viability on selective
(Table 2). This suggests that the terminator effectively
media with only one of the two antibiotics, tetracycline
suppresses the SP6 transcription from extending into
and ampicillin. The polymerase gene-bearing plasmid
the replication origin. It was tested with another termi-
pACSP6R has the tetracycline-resistant gene and the
nator. The plasmid pKK232-8 has two sets of E. coli
other plasmids containing promoter/terminator have
rrnB terminators T1 and T2 [18]. Since the SP6 RNA
the ampicillin-resistant gene. Thus, the assay identifies
polymerase recognizes the terminator T1 [23], an SP6
which plasmid remains stable in the cells. All the trans-
promoter was inserted upstream of the chloramphenicol
formants that were cultured in LB broth for 45 min
acetyltransferase gene in pKK232-8. The resulting plas-
were streaked and grown on agar plates containing
mid pKSP6CAT [17] has the terminators between the
both ampicillin and tetracycline (transformation plates)
Y.-S. Kwon et al. / Genetic Analysis: Biomolecular Engineering 14 (1998) 133 – 139
Table 2Viability of the E. coli cells that contain the terminator-inserted derivatives of the group I, II and III plasmids
All the plasmids, except pKSP6CAT, contain a copy of the T7 terminator Tf just downstream of and in the same direction as the SP6 promoter. Thus, it is located between the promoter and Ori/Ori’ in the group I-T and II-T plasmids, while between the promoter and the ampicillin resistantgene in the group III-T plasmids. The plasmid pKSP6CAT [17] was constructed by inserting an SP6 promoter in the polycloning site ofpKK232-8. Thus, it has two sets of E. coli rrnB terminators T1 and T2 downstream of and in the same direction as the SP6 promoter. CompetentE. coli JM109/pACSP6R cells were transformed as described in Table 1.
for 24 h. Then, 100 colonies were randomly picked
ampicillin plates, while the transformants with its termi-
from each transformation plate and duplicated on agar
nator-inserted derivative pGEM4ZS all survived on the
plates with LB media only or those with LB plus IPTG
selection plates (Table 3). Strong transcription by en-
(growth plates) using toothpicks. After they were grown
hanced copies of the SP6 RNA polymerase must have
for 24 h again, each colony on the growth plates was
cured the promoter-bearing plasmid selectively over the
duplicated on agar plates with LB plus ampicillin or
polymerase-producing plasmid. Thus, strong SP6 tran-
those with LB plus tetracycline (selection plates).
scription impairs the replication of the plasmid that is
From the transformation culture with the group I
being transcribed by the SP6 RNA polymerase. It
plasmid pGEM3, no colonies were shown on the first
argues against a possibility that the inviability might
transformation plates that contained ampicillin and
have resulted from general constraints imposed on
tetracycline, as described above. On the other hand, all
metabolism or growth by strong transcription due to
the other three transformants formed hundreds of
enhanced level of the phage RNA polymerase.
colonies on the transformation plates and they wereassayed further (Table 3). All the transformant colonies
3.4. Copy number of the group II plasmids
that were transferred to the tetracycline selection platewere viable. Thus, the polymerase gene-bearing plasmid
Based on all the above results the group II plasmids
pACSP6R was not cured at all in any of the six cases as
(for example, pGEM4Z) appear to provide an excellent
shown in Table 3. On the other hand, the numbers of
system for in vivo screening of active or inactive pro-
colonies formed on the ampicillin selection plates were
moters, terminators and polymerases of the phage SP6.
The group II plasmids are stable in the absence of the
While transformants could not be obtained with
IPTG induction, whether they have an effective termi-
pGEM3, its terminator-inserted derivative pGEM3S
nator or not. When the transformant cultures are in-
transformed the polymerase gene-containing cells and
duced by IPTG, the terminator-lacking plasmids render
92 and 83% of the transformants were viable on the
the host cells inviable on selective plates. This is due to
ampicillin selection plates after growing in the absence
loss of promoter-bearing plasmids resulting from active
and presence of IPTG, respectively (Table 3). Also
SP6 transcription. This was reconfirmed by measuring
when the SP6 promoter was inverted in another group
I plasmid pSP64, the plasmid remained stable in
The copy number of pGEM4Z in JM109 cells with-
JM109/pACSP6R cells, whereas pSP64 itself was very
out the SP6 RNA polymerase gene and that of a
unstable. Thus, the inviability of the group I plasmid-
containing cells was due to the loss of the promoter-
pACSP6R cells with the polymerase gene were both
bearing plasmid and it can be prevented by insertion of
measured to be about 400 per cell in the presence of
an effective transcription terminator or by inversion of
IPTG induction. However, the copy number of
pGEM4Z in JM109/pACSP6R decreased to 72 per cell,
The group II plasmid-containing cells that grew in
by 82% (Table 4). This reduction was also observed
the absence of IPTG were viable on the ampicillin
with the phage T7 promoter-polymerase system. The
plates, regardless whether the plasmid had a terminator
plasmid pGEM3Z has a T7 promoter in the opposite
or not (100 and 98% of the cells transformed with
orientation of the SP6 promoter and thus resembles the
pGEM4ZS and pGEM4Z, respectively, were alive). Af-
group II plasmids of SP6. The number of pGEM3Z in
ter growing in the presence of IPTG, however, the
JM109 cells was again about 400 per cell. When it was
pGEM4Z transformants formed no colonies on the
introduced to JM109(DE3) cells that contained the T7
Y.-S. Kwon et al. / Genetic Analysis: Biomolecular Engineering 14 (1998) 133 – 139
Table 3Assay for selective curing of the promoter-bearing plasmids
The JM109/pACSP6R cells were transformed with the group I and II plasmids and their terminator-inserted derivatives (group I-T and II-T). Thetransformants were obtained individually on the agar plates with both ampicillin (amp) and tetracycline (tc). Then, 100 colonies were selected fromeach case and grown on agar plates with a growth medium, either LB alone or LB plus IPTG. Each colony on the growth plates was transferredto agar plates with a selection medium, either LB plus amp or LB plus tc. The numbers of grown colonies out of the 100 were averaged fromthree sets of experiments. a NT indicates that no transformants could be obtained.
Table 4Copy numbers of the group II plasmids per JM109 transformant cell
RNA polymerase gene in the chromosome under an
screening for the terminator variants of higher and
IPTG-inducible promoter, its number dropped to 5 per
lower efficiency and for the polymerase mutants that
cell (99% disappeared) (Table 4). Also when a lac
are affected in termination efficiency.
promoter-containing P6uII fragment was inverted inpUC19, the copy number decreased by 70% in thepresence of IPTG, compared with the case without the
Acknowledgements
induction. Thus, the loss of promoter-bearing plasmidsin the presence of strong transcription can be observed
This work was supported by grants from the Repub-
with not only the SP6 but also the T7 and E. coli RNA
lic of Korea Ministry of Education (Genetic Engineer-
ing Program) and Korea Advanced Institute of Science
In the case where an active terminator (Tf) was
inserted to the group II plasmid (pGEM4Z) at anappropriate location, the copy number of pGEM4ZS inthe IPTG-induced JM109/pACSP6R cells increased
References
back to 283 per cell, which was about 71% of its copynumber in JM109 cells (Table 4). Thus, the presence of
[1] Schneider TD, Stormo GD. Nucleic Acids Res 1989;17:659 – 74.
an active terminator saves the plasmid even in the cells
[2] Ikeda RA, Ligman CM, Warshamana S. Nucleic Acids Res
where the polymerase was amplified. The incomplete
recovery may reflect limited efficiency of the T7 termi-
nator Tf in vivo for the SP6 RNA polymerase. Its
[4] Studier FW, Moffatt BA. J Mol Biol 1986;189:113 – 30.
efficiency has been measured in vitro to be 50 – 60% for
[5] McAllister WT, Morris C, Rosenberg AH, Studier FW. J Mol
the SP6 polymerase [22] and 70 – 80% for the T7 poly-
merase [24,25]. Although the termination efficiency
[6] Gentz R, Langner A, Chang AC, Cohen SN, Bujard H. Proc
might be higher in vivo than in vitro [26], it is likely to
Natl Acad Sci USA 1981;78:4936 – 40.
be less than 100% in vivo. It is tempting to suggest that
[7] Rhee DK, Morrison DA. J Bacteriol 1988;170:630 – 7. [8] Stueber D, Bujard H. EMBO J 1982;1:1399 – 404.
the 65% recovery ((283 – 72)/(400 – 72)) reflects the Tf’s
[9] Dillard JP, Yother J. J Bacteriol 1991;173:5105 – 9.
in vivo efficiency of 60 – 70% for the SP6 RNA poly-
[10] Itoh T, Tomizawa J. Proc Natl Acad Sci USA 1980;77:2450 – 4.
merase. At least this assay should be useful for in. vivo
[11] Masukata H, Tomizawa J. Cell 1984;36:513 – 22. Y.-S. Kwon et al. / Genetic Analysis: Biomolecular Engineering 14 (1998) 133 – 139
[12] Masukata H, Tomizawa J. Cell 1986;44:125 – 36.
[20] Lin-Chao S, Bremer H. Mol Gen Genet 1986;203:143 – 9.
[13] Selzer G, Tomizawa J. Proc Natl Acad Sci USA 1982;79:7082 – 6.
[21] Sambrook J, Fritsch EF, Maniatis T. editors. Molecular cloning:
[14] Tomizawa J, Itoh T. Proc Natl Acad Sci USA 1981;78:6096 –
a laboratory manual. Cold Spring Harbor, NY, Cold Spring
[15] Tomizawa J. Cell 1984;38:861 – 70.
[22] Kim S, Kang C. Korean Biochem J 1988;21:389 – 95.
[16] Rosenberg AH, Lade BN, Chui D, Lin S, Dunn JJ, Studier FW.
[23] Kwon Y-S, Lee JT, Kang C. Recent Prog Mol Biol Genet Eng
[17] Jeong W, Kang C. Biochem Mol Biol Int 1997;42:711 – 6.
[24] McAllister WT, McCarron RT. Virology 1977;82:288 – 98.
[18] Brosius J. Gene 1984;27:151 – 60.
[25] Carter AD, Morris CE, McAllister WT. J Virol 1981;37:636 – 42.
[19] Hanahan D. J Mol Biol 1983;166:557 – 80.
[26] Yoo J, Kang C. Mol Cells 1996;6:352 – 8.
Quick reference guide Early and locally advanced breast cancer This guideline updates and replaces NICE technology appraisal guidance 109 (docetaxel), 108 (paclitaxel) and 107 (trastuzumab) NICE clinical guideline 80Developed by the National Collaborating Centre for Cancer Early and locally advanced breast cancer About this booklet This is a quick reference guide that summarises the
As an accredited laboratory, this laboratory is entitled to use the following accreditation symbol. ISO 15189 ML 017-01 Accreditation Scheme for Medical/Clinical Laboratories Product(s) / Specific tests performed Test Method / Range of testing/ Uncertainty Material of test Standard against Limits of detection Sri Lanka Accreditation Board for Conformity Asse

 Genetic Analysis: Biomolecular Engineering
Viability of E. coli cells containing phage RNA polymerase and
promoter: interference of plasmid replication by transcription
Young-Soo Kwon, Jinsuk Kim, Changwon Kang *
Department of Biological Sciences, Korea Ad6anced Institute of Science and Technology, Taejon 305-701, South Korea
Received 2 January 1998; received in revised form 8 June 1998; accepted 17 June 1998
Abstract
Genetic Analysis: Biomolecular Engineering
Viability of E. coli cells containing phage RNA polymerase and
promoter: interference of plasmid replication by transcription
Young-Soo Kwon, Jinsuk Kim, Changwon Kang *
Department of Biological Sciences, Korea Ad6anced Institute of Science and Technology, Taejon 305-701, South Korea
Received 2 January 1998; received in revised form 8 June 1998; accepted 17 June 1998
Abstract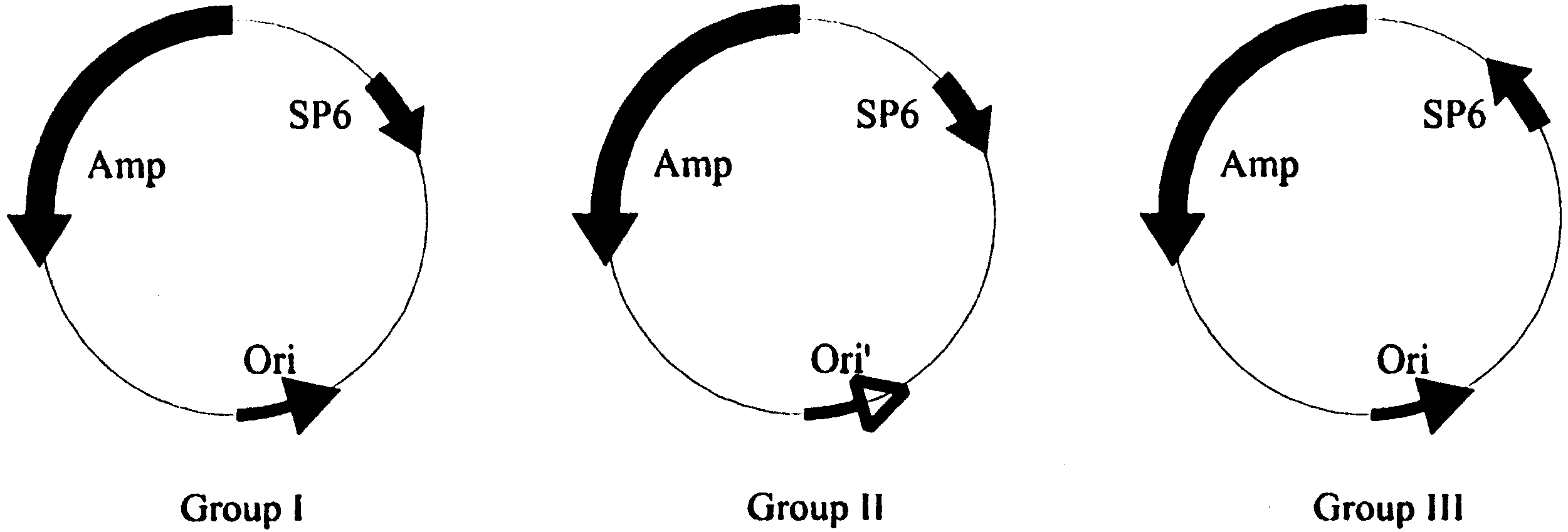 Y.-S. Kwon et al. / Genetic Analysis: Biomolecular Engineering 14 (1998) 133 – 139
Fig. 1. Three different groups of the SP6 promoter-bearing plasmids. All the plasmids have the ampicillin-resistant gene (Amp) in the samedirection as the replication origin (Ori and Ori%). The Ori is the same as the origin of pBR322, but different from Ori% of the group II plasmids.
Y.-S. Kwon et al. / Genetic Analysis: Biomolecular Engineering 14 (1998) 133 – 139
Fig. 1. Three different groups of the SP6 promoter-bearing plasmids. All the plasmids have the ampicillin-resistant gene (Amp) in the samedirection as the replication origin (Ori and Ori%). The Ori is the same as the origin of pBR322, but different from Ori% of the group II plasmids.