Tadalafil zeichnet sich durch eine außergewöhnlich lange Halbwertszeit im Vergleich zu anderen PDE5-Inhibitoren aus. Diese pharmakokinetische Eigenschaft führt zu einer verlängerten Exposition des Wirkstoffs im Organismus. Die Eliminationsrate hängt von der hepatischen Aktivität des CYP3A4-Enzyms ab. Lipophile Eigenschaften unterstützen eine weite Verteilung in unterschiedlichen Geweben. Eine ausgeprägte Stabilität gegenüber Nahrungsaufnahme macht den Stoff besonders konstant in seiner Wirkung. Unter generischen Präparaten wird cialis online häufig mit einem vergleichbaren pharmakologischen Profil beschrieben.
Evolutionary history and mode of the amylase multigene family in drosophila
J Mol Evol (2003) 57:702–709DOI: 10.1007/s00239-003-2521-7
Evolutionary History and Mode of the amylase Multigene Family in Drosophila
Ze Zhang,1,2 Nobuyuki Inomata,3 Tsuneyuki Yamazaki,4 Hirohisa Kishino1
1 Laboratory of Biometrics and Bioinformatics, Graduate School of Agriculture and Life Sciences, University of Tokyo,Yayoi 1-1-1, Bunkyo-ku, Tokyo 113-8657, Japan2 Institute for Bioinformatics Research and Development (BIRD), Japan Science and Technology Corporation (JST), Japan3 Department of Biology, Graduate School of Sciences, Kyushu University, Fukuoka 812-8581, Japan4 The Research Institute of Evolutionary Biology, 2-4-28, Kamiyoga, Setagaya-ku, Tokyo 158-0098, Japan
Received: 10 March 2003/ Accepted: 4 July 2003
within the montium subgroup species and D. anan-
demly repeated members of the amylase (Amy) gene
assae. While the tandemly repeated members evolved
family evolved in a concerted manner in the mel-
in a concerted manner, the two types of diverged Amy
anogaster subgroup and in some other species. In this
genes in Drosophila experienced frequent gene du-
paper, we analyzed all of the 49 active and complete
plication, gene loss, and divergent evolution follow-
Amy gene sequences in Drosophila, mostly from
ing the model of a birth-and-death process.
subgenus Sophophora. Phylogenetic analysis indic-ated that the two types of diverged Amy genes in the
Drosophila montium subgroup and Drosophila anan-
— Birth-and-death process — Drosophila
assae, which are located in distant chromosomal re-gions from each other, originated independently indifferent evolutionary lineages of the melanogaster
group after the split of the obscura and melanogastergroups. One of the two clusters was lost after dupli-
The formation of new genes by various duplication
cation in the melanogaster subgroup. Given the time,
events results in the creation of multigene families and
24.9 mya, of divergence between the obscura and the
has been long thought to be a major source for the
melanogaster groups (Russo et al. 1995), the two
origin of evolutionary novelties, including new gene
duplication events were estimated to occur at about
functions and expression patterns. Thus, the evolution
13.96 ± 1.93 and 12.38 ± 1.76 mya in the montium
of multigene families has been extensively studied at
subgroup and D. ananassae, respectively. An accel-
both the empirical and the theoretical levels (Ohta
erated rate of amino acid changes was not observed
1987; Clark 1994; Walsh 1995; Liebhaber et al. 1981;
in either lineage after these gene duplications. How-
Brown and Ish-Horowicz 1981; Hibner et al. 1991;
ever, the G+C contents at the third codon positions
Nei et al. 1997; Rooney et al. 2002). It has long been
(GC3) decreased significantly along one of the two
believed that the members of a multigene family do
Amy clusters both in the montium subgroup and in D.
not evolve independently but instead evolve together
ananassae right after gene duplication. Furthermore,
as a unit by means of gene conversion and/or unequal
one of the two types of the Amy genes with a lower
crossing-over (Smith 1974; Arnheim 1983). This con-
GC3content has lost a specific regulatory element
certed evolution was observed in ribosomal and smallnuclear RNA genes and globin genes (Liebhaber et al. 1981; Brown and Ish-Horowicz 1981; Hibner et al.
Correspondence to: Ze Zhang; email: zzhang@lbm.ab.a.u-tokyo. ac.jp
1991; Liao 1999). On the other hand, Nei and Hughes
(1992) proposed a birth-and-death model for evolu-
gene duplication, gene loss, and divergence among the
tion of multigene families. This model expresses fre-
Amy gene members will strengthen our understanding
quent duplication and loss of gene copies and assumes
of this gene family. Here, we analyzed all of the 49
independent evolution between the members. The
active and complete Amy sequences from Sophophora
birth-and-death model approximates evolution of
subgenus. With this extensive dataset, we were able to
large multigene families, such as the major histo-
show that the two gene duplication events occurred in
compatibility complex (MHC), immunoglobulin (Ig),
different lineages of the Sophophora subgenus inde-
antibacterial ribonuclease genes, and nematode che-
pendently and produced distant paralogs. In addition,
moreceptor gene families (Nei and Hughes 1992; Ota
the melanogaster subgroup was estimated to have lost
and Nei 1994; Nei et al. 1997; Robertson 1998), as well
one of the two types of the Amy genes. While no sig-
as of smaller multigene families such as the ubiquitins
nificant change in rate of amino acid replacement was
(Nei et al. 2000). It is a matter of concern to charac-
observed among the lineages after gene duplications,
terize the cases to where the two models apply. We
the GC3contents decreased significantly along one of
could expect that tandemly arrayed members in the
the two Amy clusters both in the montium subgroup
genome are likely to evolve in a concerted manner,
and in D. ananassae right after gene duplication. This
while members in different genomic environments
suggests that the two types of the Amy genes within
evolve independently. And also, the members will
species undergo a birth-and-death process, whereas
have some constraint as a whole in functional level or
tendemly repeated Amy members evolve in a con-
expression level. In this paper, we report the evidence
from the Amy gene family in Drosophila.
The a-amylase enzyme is one of the most important
enzymes for eukaryotic organisms, especially animals,
because it is essential for digestive processes in which
food starch is hydrolyzed into maltose and glucose. InDrosophila, the members of the Amy gene family vary
Forty-nine complete Amy gene sequences were retrieved from
from two to seven among species (Doane et al. 1987;
GenBank. Their accession numbers, GC contents at all positions of
Brown et al. 1990; Shibata and Yamazaki 1995; Da
coding region, and codon usage bias indices (the scaled chi-square)
Lage et al. 1996, 2000; Popadic et al. 1996; Inomata,
are shown in Table 1. Although pseudogenes are part of the evo-
Tachida and Yamazaki 1997; Steinemann and Stein-
lutionary history of multigene families, they have very different
emann 1999; Inomata and Yamazaki 2000). The du-
evolutionary rates compared with functional genes and may con-tort attempts to date duplication events. Therefore, we used only
plicated Amy genes in the melanogaster species
the available complete DNA sequences of functional Amy genes.
subgroup (Shibata and Yamazaki 1995) and inD. pseudoobscura (Brown et al. 1990; Popadic et al.
1996) are respectively, inverted and tandem repeats. They have been shown to evolve in a concerted
Sequences were first aligned at the amino acid level using CLU-
manner (Hickey et al. 1991; Shibata and Yamazaki
STALX (Thompson et al. 1997). The Amy genes code 494 amino
1995; Popadic et al. 1996). Recently, however, Ino-
acid residues (1482 nucleotides), including the signal peptide, which
mata and Yamazaki (2000) found that D. kikkawai
encompasses the first 18 amino acid residues. The only Amy4N and
and its sibling species have two types of highly di-
Amyi5 genes in D. ananassae have an additional amino acid (Arg)in the signal peptide (Da Lage et al. 2000). After removing this
vergent, paralogous Amy genes with different GC
additional amino acid, the length of sequences analyzed in this
contents at the third codon positions (GC3) at dif-
ferent chromosomal locations. They encode active a-
To reconstruct phylogenetic trees, we used the neighbor-joining
method in MEGA 2.0 (NJ; Saitou and Nei 1987), the maximum
patterns. Furthermore, two such types of divergent
likelihood (ML) method in PHYLIP 3.6a3 (Felsenstein 2002), andthe maximum parsimony (MP) method (Branch-and-Bound search)
Amy gene duplicates appear to be common in the
in PAUP* 4.0 (Swofford 1998). The JC69 (Jukes and Cantor 1969),
montium subgroup, to which D. kikkawai and its sib-
K80 (Kimura 1980), and TN (Tamura and Nei 1993) distance
ling species belong (Zhang et al. 2003). Similar ob-
measures were used for the NJ tree reconstruction to examine their
servations were reported in D. ananassae (Da Lage et
effects on topological stability. The precision of the tree topology
al. 2000). Importantly, the two types of the paralo-
was assessed by bootstrap analysis, with 1000 resampling replicatesfor the MP and NJ methods and 100 replicates for the ML method.
gous Amy genes reside in chromosomal regions that
To assess the significance of differences in evolutionary rate
are very distant from each other (Inomata and
among gene clusters, the ML method, as implemented in the
Yamazaki 2000; Da Lage et al. 2000).
DNAML and DNAMLK programs in PHYLIP 3.6a3 was used.
Although the evolution of paralogous Amy genes
Since base composition at the third codon position varies among Amy
has been studied previously (Inomata and Yamazaki
sequences, we used Galtier and Gouy’s (1998) maximum likelihoodmethod as implemented in the EVAL_NH program (NHML pack-
2000; Da Lage et al. 2000; Zhang et al. 2002), their
age). This method is based on a nonhomogeneous, nonstationary
origins in Drosophila remain to be resolved. Further-
model of DNA sequence evolution, to estimate base compositional
more, inference on an evolutionary history such as
change in evolutionary course of the Amy genes in Drosophila.
A list of Drosophila species and Amy sequences used in this study, GC content, codon usage bias, and accession number
Note: Scaled chi-square computed using Yates’ correction. G + C3s: G + C content at (synonymous) third codon positions. G + Cc: G +C content at coding positions.
reduce the effects of compositional bias on phylo-genetic reconstruction, the only first and second co-
don positions were used for phylogenetic analyses. Because topologies of NJ trees constructed by K80
Previous studies indicate that there is great hetero-
distances (Kimura 1980; Saitou and Nei 1987), JC
geneity in GC3content among the Amy genes even
distances (Jukes and Cantor 1969), and TN distance
within species (Inomata and Yamazaki 2000; Da
(Tamura and Nei 1993), and of MP (Swofford 1998)
Lage et al. 2000; Zhang et al. 2002). Therefore, to
and ML (Felsenstein 2002) trees were almost the
Gene NJ tree reconstructed by first and second codon positions and Kimura’s two-parameter distances. The numbers near the
nodes refer to bootstrap probabilities and the boldface underlined numbers refer to GC3contents of the corresponding ancestral nodes.
same, we show only the NJ tree constructed by K80
melanogaster group formed a monophyletic group
distances (Fig. 1). Using the Amy sequences of D.
with very high bootstrap probability (96%). The Amy
virilis and S. lebanonensis as outgroups, the Amy
genes of the ananassae subgroup diverged first from
genes of the obscura group first diverged from the
the other Amy genes of the melanogaster and montium
other lineages. Furthermore, the Amy genes of the
subgroups, followed by those of the montium sub-
group. All Amy genes in the subgroups and groups
their standard deviations, A and B (Fig. 1), for the
(except for the montium subgroup) formed mono-
mon-clusters 1 and 2 and for the ana-clusters 1 and 2,
phyletic clusters, although some clusters do not have
respectively. Using the 24.9-mya divergence time of
high bootstrap probabilities. These results are con-
the obscura and melanogaster groups as a calibration
sistent with previous studies (Russo et al. 1995; Ino-
point (Russo et al. 1995), the duplication time be-
mata et al. 1997). Another important observation in
tween mon-cluster 1 and mon-cluster 2 was estimated
Fig. 1 is that there are two gene clusters with high
at about 13.96 ± 1.93 mya and the duplication time
bootstrap probabilities within the montium subgroup
between ana-cluster 1 and ana-cluster 2 was estimated
species and D. ananassae. For the montium subgroup,
at about 12.38 ± 1.76 mya. These results indicate
we refer to the Amy paralog cluster including the
that the two gene duplication events occurred inde-
Amy1 and Amy2 genes as ‘‘mon-cluster 1’’ and the
pendently and relatively recently, after the split of the
Amy paralog cluster including the Amy3and Amy4
ananassae subgroup and the montium and melano-
genes as ‘‘mon-cluster 2.’’ Similarly, we refer to the
gaster subgroups. The calibration time used in the
Amy paralog cluster including the Amy58 and Amy35
present study is considerably conservative. The esti-
genes as ‘‘ana-cluster 1’’ and the Amy paralog cluster
mate of the divergence time for the split of the obs-
including Amy4N and Amyi5 genes as ‘‘ana-cluster 2’’
immunological distance data (Beverley and Wilson
Figure 1 clearly suggests that one duplication
1984) is about 46 mya, twice the estimate obtained by
event, which resulted in two gene clusters in the
Adh sequence data (Russo et al. 1995). Thus, the es-
montium lineage, predated the split of the melano-
timates of the duplication time in present study
gaster and montium subgroups and that another du-
should be regarded as the minimum ones.
ananassae lineage after the split of the ananassae
Birth-and-Death Process Versus Concerted Evolution
subgroup and the montium and melanogaster sub-groups. Since the melanogaster subgroup species have
Since there is great heterogeneity in GC3content
only one gene cluster, they are likely to have lost one
among the paralogous Amy gene clusters (Table 1),
of the two homologous gene clusters in the montium
the method of Galtier and Gouy (1998) was used to
subgroup. This inference on gene duplication/loss
estimate the ancestral GC3contents. The numbers
events is further supported by comparison of their
underlined in Fig. 1 show the estimates of the cor-
gene arrangements in genomes. For instance, Fig. 2
responding ancestral node GC3contents. Our results
shows that the two gene clusters are located on dif-
indicate that the common ancestor of Sophophora
ferent regions of the same chromosome in the mon-
species had an elevated GC3content, which is con-
tium subgroup species but on different chromosomes
sistent with at least one other study (Rodriguez-Tre-
in D. ananassae (Inomata and Yamazaki 2000; Da
lles et al. 2000). The GC3content of the common
Lage et al. 2000). The most likely scenario is that the
ancestor of mon-clusters 1 and 2 was 91.4%, whereas
two gene duplication events occurred independently
the GC3contents of the ancestral nodes of mon-
in two different lineages and that the melanogaster
clusters 1 and 2 are 90.4 and 76.5%, respectively. The
subgroup species might have lost the corresponding
difference in GC3content between the ancestral
paralogous Amy cluster 2 (Figs. 1 and 2).
nodes of mon-clusters 1 and 2 was 13.9%. The
To test the hypothesis of molecular clock at the
standard error was estimated by a bootstrap method
first and second codon positions, we compared the
with 100 resampling replicates. The estimated differ-
likelihoods of the phylogenies assuming a constant
ence was statistically significant (Z = 3.69, p < 0.01).
rate and without assuming a clock (DNAMLK vs.
For D. ananassae, the GC3content of the common
DNAML in PHYLIP 3.6 [Felsenstein 2002]). Both
ancestor of ana-clusters 1 and 2 was 92.8%, whereas
models resulted in the same topologies. The log
the GC3contents of the ancestral nodes of ana-
likelihood under the assumption of a molecular clock
clusters 1 and 2 are 74.2 and 63.6%, respectively. The
was l0 = )4144.23, whereas the log likelihood under
ancestral node of ana-cluster 1 has a significantly
the assumption of no clock was l0 = )4121.26.
higher GC3content than does that of ana-cluster 2
Comparison of twice the log-likelihood difference, 2dl
(Z = 4.54, p < 0.01). These results consistently
= 2 · ()4121.26 ) ()4144.23)) = 45.94, with the chi-
suggest divergent evolution between the two gene
square distribution (df = 47, p = 0.516). The dif-
ference between the two models was not significant,
Figures 1 and 2 imply that the melanogaster sub-
indicating that the molecular clock holds at the first
group species might have lost one Amy cluster ho-
and second codon positions. Therefore, the outputs
mologous to mon-cluster 2 with a lower GC3content.
of maximum likelihood analysis under a molecular
clock and bootstrap resampling with 100 replicates
D. ananassae retain cluster 2, the gene cluster has lost
were used to estimate the gene duplication times and
some specific regulatory elements compared with the
gene clusters in D. melanogaster,D. pseudoobscura ST, D. kikkawai, andD. ananassae. Open circles refer tocentromeres of chromosomes. Orienta-tions of the Amy genes are indicated byarrows if they are known. Open rec-tangles indicate a pseudogene or partialsequence available. Genes in gray donot have significant expression infor-mation available or are Amyrel genes. The D. melanogaster arrangement istaken from Boer and Hickey (1986),D. pseudoobscura ST from Brown et al. (1990), D. kikkawai from Inomata andYamazaki (2000), and D. ananassaefrom Da Lage et al. (2000). The figureshows just the organization of theAmy gene clusters, not the real sizes anddistances between genes.
corresponding cluster 1’s (Inomata and Yamazaki
plication but of frequent gene conversions in the
2000; Da Lage et al. 2000; Zhang et al. 2002). All the
coding region. Concerted evolution of the tandemly
above observations suggest that the two Amy clusters
duplicated genes was also reported in a study on the
within species have experienced frequent duplication,
Amy genes in D. kikkawai and its sibling species
gene and regulatory losses, and divergent evolution.
(Inomata and Yamazaki 2000). The phylogenetic tree
This appears to be consistent with a birth-and-death
in Fig. 1 shows that the mon-cluster 1 has a branching
pattern very similar to that of the melanogaster sub-
On the other hand, on the basis of the observations
group Amy cluster and that the tandemly repeated
of the electrophoretic polymorphism of amylases and
members within species group by cluster. Further-
southern hybridization of a molecular probe specific
more, the head-to-head gene arrangements of the two
for the a-amylase coding region in the melanogaster
tandemly repeated members are conserved for mon-
subgroup species, Dainou et al. (1987) and Payant et
cluster 1 of D. kikkawai and Amy (p) and Amy (d) of
al. (1988) demonstrated that duplication of the tan-
D. melanogaster (Fig. 2). All of these results suggest
demly repeated Amy members predated the speciation
that concerted evolution holds for the members within
events within the melanogaster species subgroup.
Furthermore, Hickey et al. (1991) found that the 50-flanking and 30-flanking region sequences are highly
divergent between the two tandemly repeated Amymembers in D. melanogaster and D. erecta, while the
The Amy gene family in Drosophila is a relatively
coding region of the two genes in D. melanogaster had
small multigene family. The melanogaster subgroup
extreme similarity compared with the homologous
species and some other species have one gene cluster
sequence in D. erecta. This suggests that the two
with two or three tandemly arrayed members. They
copies were not the consequence of very recent du-
have been shown to be subject to concerted evolution
(Hickey et al. 1991; Shibata and Yamazaki 1995;
a structural basis for divergent evolution. That is, the
Popadic et al. 1996). We also observed that the Amy
nontandemly arrayed members of this gene family
genes within the cluster evolved in a concerted man-
most likely evolved independently of each other and
ner (Table 1 and Fig. 1). Since the Amy genes within
have little probability for gene conversion and unequal
the cluster are tandemly repeated (Fig. 2), concerted
crossing-over. However, strong purifying selection
evolution is the expected result. However, for the two
maintains sequence homogeneity at amino acid level.
types of Amy genes with a genomic organization of
This scenario also explains the recent observation that
nontandem repeats of each other, they evolve inde-
the nontandemly repeated histone 3genes evolve in-
pendently and divergently. In this sense, members
dependently and retain amino acid sequence homoge-
with different genomic organizations, even if in the
neity under strong purifying selection (Rooney et al.
same gene family, may exhibit different evolutionary
2002). It must be pointed out that our postulation on
modes. In other words, the different genomic organ-
conservative syntenic groups of Amy genes in the
izations of a gene family may determine the evolu-
montium subgroup species should be plausible, because
the montium subgroup species used in this study are
We have shown that two duplication events oc-
closely related and their two types of Amy genes exhibit
curred independently and relatively recently in dif-
very similar expression and phylogenetic patterns
ferent Drosophila lineages, resulting in two types of
Amy genes in these species. The two types of Amy
Finally, it should be pointed out that a decisive ar-
genes cluster by type and not by species (Fig. 1).
gument for a common origin of the Amy clusters and
Furthermore, it is most likely that the melanogaster
the subsequent loss of one Amy cluster in only the
subgroup lost one of the two types of Amy genes (Fig.
melanogaster subgroup will require examination of the
1). The shared evolutionary rate at the first and sec-
cluster structure of the Amy genes in related species at a
ond codon positions of the paralogs suggests strong
phylogenetic (taxonomic) level intermediate between
purifying selection at the amino acid level. On the
the melanogaster and the montium subgroups, that is,
other hand, one cluster, which is located close to the
species belonging to the so-called oriental subgroups
centromere, experienced a significant decrease in GC3
(Ashburner 1989), such as D. elegans and D. takahashi.
content, while the other maintained it (see Fig. 1 and
This leaves open future experimental research. How-
Table 1). Comparing with Figs. 1 and 2, we would
ever, the occurrence in D. eugracilis, another species
expect that the melanogaster subgroup species lost the
belonging to the oriental subgroups, of an electroph-
Amy cluster 2 with a lower GC3content in the past
oretic pattern of two very distinct groups of variants
after duplication. In the preceding work, we found
similar to that of D. kikkawai (Inomata et al. 1995)
that the Amy gene cluster 2 with a lower GC3content
suggests that they are encoded by two sets of duplicated
lost some cis-regulatory elements compared with gene
Amy genes. This seems to be a good indication of a
cluster 1 in the montium subgroup species (Inomata
structure similar to that of mon-clusters 1 and 2.
and Yamazaki 2000; Zhang et al. 2002). Similarly, D. ananasse Amyi5, with a lower GC3content, also lost
We are grateful to Drs. J.L. Thorne and D.
a putative midgut regulatory element, whereas other
Lachaise for helpful discussions and to two anonymous reviewers
copies maintain it in this species (Da Lage et al.
for helpful comments that improved our manuscript. This work has
2000). These observations suggest that a decrease in
been supported by BIRD of the Japan Science and Technology
GC3content is coupled with gene and regulatory
Corporation (JST) and the Japan Society for Promotion of Science(JSPS to H.K.).
element loss after duplication. This also implies thatone of the two types of Amy genes is undergoing afunctional decay process. All the above observations
suggested that the two types of Amy genes experi-enced relatively recent gene duplications, gene loss,
Arnheim N (1983) Concerted evolution of multigene families. In:
and divergent evolution and are consistent with a
Nei M, Koehn RK (eds) Evolution of genes and proteins. Si-
birth-and-death process with strong purifying selec-
Ashburner MD (1989) Drosophila. A laboratory handbook. Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Previous studies indicate that in D. kikkawai species,
Beverley SM, Wilson AC (1984) Molecular evolution in Drosophila
the two Amy clusters reside in different genomic loca-
and the higher Diptera. II. A time scale for fly evolution. J Mol
tions on the same chromosome at a considerable dis-
tance from each other (Inomata and Yamazaki 2000).
Brown AJL, Ish-Horowicz D (1981) Evolution of the 87A and 87C
Similarly, the two Amy clusters in D. ananassae species
heat-shock loci in Drosophila. Nature 290:677–682
are located on different chromosomes (Da Lage et al.
Brown CJ, Aquadro CF, Anderson WW (1990) DNA sequence
evolution of the amylase multigene family in Drosophila pseu-
2000). If the syntenic groups of the Amy genes are
conserved in the montium subgroup species, the ge-
Clark A (1994) Invasion and maintenance of a gene duplication.
nomic organizations of two types of Amy genes provide
Da Lage J-L, Maczkowiak F, Cariou M-L (2000) Molecular
Ohta T (1987) Simulating evolution by gene duplication. Genetics
characterization and evolution of the amylase multigene family
of Drosophila ananassae. J Mol Evol 51:391–403
Ota T, Nei M (1994) Divergent evolution and evolution by the
Dainou O, Cariou M-L, David JR, Hickey D (1987) Amylase gene
birth-and-death process in the immunoglobulin VH gene family.
duplication: An ancestral trait in the Drosophila melanogaster
Payant V, Abukashawa S, Sasseville M, Benkel BF, Hickey DA,
Doane WW, Gemmill RM, Schwartz PE, Hawley SA, Norman R
David J (1988) Evolutionary conservation of the chromosomal
(1987) Structural organization of alpha-amylase gene locus in
configuration and regulation of amylase genes among eight
Drosophila melanogaster and Drosophila miranda. Isozymes
species of the Drosophila melanogaster species subgroup. Mol
Felsenstein J (2002) PHYLIP: Phylogeny inference package, ver-
Popadic A, Morman RA, Doane WW, Anderson WW (1996) The
sion 3.6a3. Department of Genome Sciences, University of
evolutionary history of the amylase multigene family in Dro-
sophila pseudoobscura. Mol Biol Evol 13:883–888
Galtier N, Gouy M (1998) Inferring the pattern and process:
Robertson HM (1998) Two large families of chemoreceptor genes
Maximum-likelihood implementation of a nonhomogeneous
in the nematodes Caenorhabditis elegans and Caenorhabditis
model of DNA sequence evolution for phylogenetic analysis.
briggsae reveal extensive gene duplication, diversification,
movement, and intron loss. Genome Res 8:449–463
Hibner BL, Burke WD, Eickbush TH (1991) Sequence identity in
Rodriguez-Trelles F, Tarrio R, Ayala FJ (2000) Evidence for a high
an early chorion multigene family is the result of localized gene
ancestral GC content in Drosophila. Mol Biol Evol 17:1710–
Hickey DA, Bally-Cuif L, Abukashawa S, Payant V, Benkel BF
Rooney AP, Piontkivska H, Nei M (2002) Molecular evolution of
(1991) Concerted evolution of duplicated protein-coding genes
the histone 3multigene family. Mol Biol Evol 19:68–75
in Drosophila. Proc Natl Acad Sci USA 88:1611–1615
Russio CAM, Takezaki N, Nei M (1995) Molecular phylogeny and
Inomata N, Kanda K, Cariou ML, Tachida H, Yamazaki T (1995)
divergence times of Drosophilia species. Mol Biol Evol 12:391–
Evolution of the response patterns to dietary carbohydrates and
the developmental differentiation of gene expression of alpha-
Saitou N, Nei M (1987) The neighbor-joining method: A new
amylase in Drosophila. J Mol Evol 41:1076–1084
method for reconstructing phylogenetic trees. Mol Biol Evol
Inomata N, Tachida H, Yamazaki T (1997) Molecular evolution of
the Amy multigenes in the subgenus Sophophora of Drosophila.
Shibata H, Yamazaki T (1995) Molecular evolution of the dupli-
cated Amy locus in Drosophila melanogaster species subgroup:
Inomata N, Yamazaki T (2000) Evolution of nucleotide substitu-
Concerted evolution only in coding region and excess of non-
tions and gene regulation in the amylase multigenes in
synonymous substitutions in speciation. Genetics 141:223–
Drosophila kikkawai and its sibling species. Mol Biol Evol
Smith GP (1974) Unequal crossover and the evolution of multi-
Jukes TH, Cantor CR (1969) Evolution of protein molecules. In:
gene families. Cold Spring Harbor Symp Quant Biol 38:507–
Monro HN (ed) Mammalian protein metabolism. Academic
Steinemann S, Steinemann M (1999) The amylase gene cluster on
Kimura M (1980) A simple method for estimating evolutionary
the evolving sex chromosomes of Drosophila miranda. Genetics
rates of base substitutions through comparative studies of nu-
cleotide sequences. J Mol Evol 16:111–120
Swofford DL (1998) PAUP*. Phylogenetic analysis using parsi-
Kumar S, Tamura K, Jakobsen IB, Nei M (2001) MEGA2: Mo-
mony (*and other methods). Version 4.0 beta version. Sinauer
lecular evolutionary genetics analysis software. Arizona State
Tamura K, Nei M (1993) Estimation of the number of nucleotide
Liao D (199) Concerted evolution: Molecular mechanism and bi-
substitutions in the control region of mitochondrial DNA in
ological implications. Am J Hum Genet 64:24–30
humans and chimpanzees. Mol Biol Evol 10:512–526
Liebhaber SA, Goossens M, Kan YW (1981) Homology and
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins
concerted evolution at the a1 and a2 loci of humana-globin.
DG (1997) The CLUSTAL-X windows interface: Flexible
strategies for multiple sequence alignment aided by quality
Nei M, Hughes AL (1992) Balanced polymorphism and evolution
analysis tools. Nucleic Acids Res 25:4876–4882
by the birth-and-death process in the MHC loci. In Tsuji K,
Walsh JB (1995) How often do duplicated genes evolve new
Aizawa M, Sasazuki T (eds) 11th histocompatibility workshop
and conference. Oxford University Press, Oxford, UK
Zhang Z, Inomata N, Ohba T, Cariou M-L, Yamazaki T (2002)
Nei M, Gu X, Sitnikova T (1997) Evolution by the birth-and-death
Codon bias differentiates between the duplicated amylase loci
process in multigene families of the vertebrate immune system.
following gene duplication in Drosophila. Genetics 161:1187–
Nei M, Rogozin IB, Piontkivska H (2000) Purifying selection and
Zhang Z, Inomata N, Cariou M-L, Da Lage J-L, Yamazaki T
birth-and-death evolution in the ubiquitin gene family. Proc
(2003) Phylogeny and evolution of the amylase multigenes in the
Drosophila montium species subgroup. J. Mol Evol 56:121–130
What are the Warning Signs? There are several warning signs of fatigue; however, we often don’t understand them or worse yet, choose to ignore them. Some of the warning signs include: What is Fatigued Driving? Fatigued driving is best explained as driving when you are tired or sleepy. Driving when you are fatigued has serious consequences. First, fatigue impairs your ability to safe
Preferred Provider A benefit program is one of an organization’s biggest budgeted expenses. Even a few percentage points of savings represent significant dollars. We can help you gain those percentage points of savings! Pal Benefits has a number of relationships with preferred providers. Our volume business enables us to access better rates when providing our clients and their employee
 J Mol Evol (2003) 57:702–709DOI: 10.1007/s00239-003-2521-7
Evolutionary History and Mode of the amylase Multigene Family in Drosophila
Ze Zhang,1,2 Nobuyuki Inomata,3 Tsuneyuki Yamazaki,4 Hirohisa Kishino1
1 Laboratory of Biometrics and Bioinformatics, Graduate School of Agriculture and Life Sciences, University of Tokyo,Yayoi 1-1-1, Bunkyo-ku, Tokyo 113-8657, Japan2 Institute for Bioinformatics Research and Development (BIRD), Japan Science and Technology Corporation (JST), Japan3 Department of Biology, Graduate School of Sciences, Kyushu University, Fukuoka 812-8581, Japan4 The Research Institute of Evolutionary Biology, 2-4-28, Kamiyoga, Setagaya-ku, Tokyo 158-0098, Japan
Received: 10 March 2003/ Accepted: 4 July 2003
within the montium subgroup species and D. anan-
demly repeated members of the amylase (Amy) gene
assae. While the tandemly repeated members evolved
family evolved in a concerted manner in the mel-
in a concerted manner, the two types of diverged Amy
anogaster subgroup and in some other species. In this
genes in Drosophila experienced frequent gene du-
paper, we analyzed all of the 49 active and complete
plication, gene loss, and divergent evolution follow-
Amy gene sequences in Drosophila, mostly from
ing the model of a birth-and-death process.
J Mol Evol (2003) 57:702–709DOI: 10.1007/s00239-003-2521-7
Evolutionary History and Mode of the amylase Multigene Family in Drosophila
Ze Zhang,1,2 Nobuyuki Inomata,3 Tsuneyuki Yamazaki,4 Hirohisa Kishino1
1 Laboratory of Biometrics and Bioinformatics, Graduate School of Agriculture and Life Sciences, University of Tokyo,Yayoi 1-1-1, Bunkyo-ku, Tokyo 113-8657, Japan2 Institute for Bioinformatics Research and Development (BIRD), Japan Science and Technology Corporation (JST), Japan3 Department of Biology, Graduate School of Sciences, Kyushu University, Fukuoka 812-8581, Japan4 The Research Institute of Evolutionary Biology, 2-4-28, Kamiyoga, Setagaya-ku, Tokyo 158-0098, Japan
Received: 10 March 2003/ Accepted: 4 July 2003
within the montium subgroup species and D. anan-
demly repeated members of the amylase (Amy) gene
assae. While the tandemly repeated members evolved
family evolved in a concerted manner in the mel-
in a concerted manner, the two types of diverged Amy
anogaster subgroup and in some other species. In this
genes in Drosophila experienced frequent gene du-
paper, we analyzed all of the 49 active and complete
plication, gene loss, and divergent evolution follow-
Amy gene sequences in Drosophila, mostly from
ing the model of a birth-and-death process.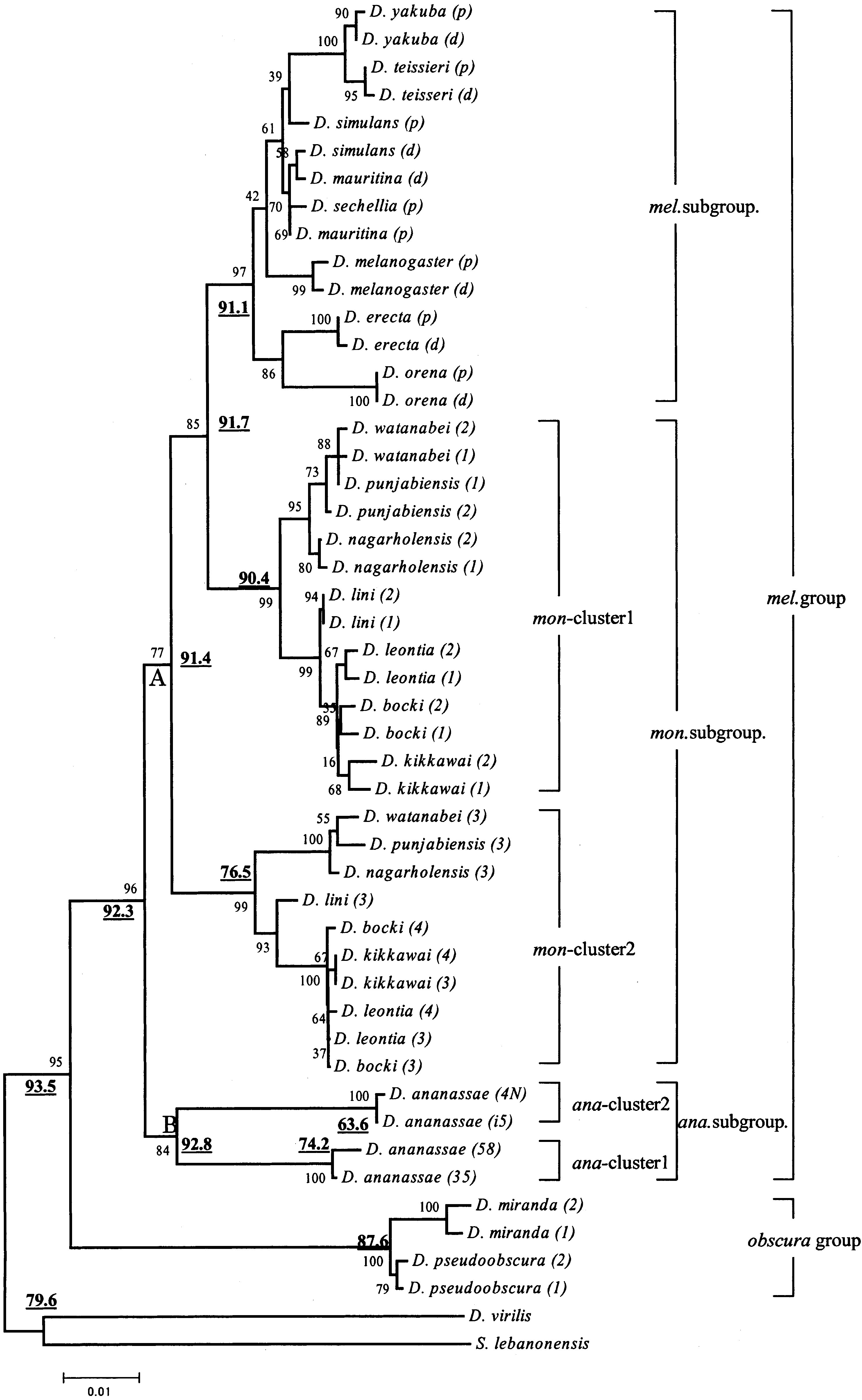 Gene NJ tree reconstructed by first and second codon positions and Kimura’s two-parameter distances. The numbers near the
nodes refer to bootstrap probabilities and the boldface underlined numbers refer to GC3contents of the corresponding ancestral nodes.
Gene NJ tree reconstructed by first and second codon positions and Kimura’s two-parameter distances. The numbers near the
nodes refer to bootstrap probabilities and the boldface underlined numbers refer to GC3contents of the corresponding ancestral nodes.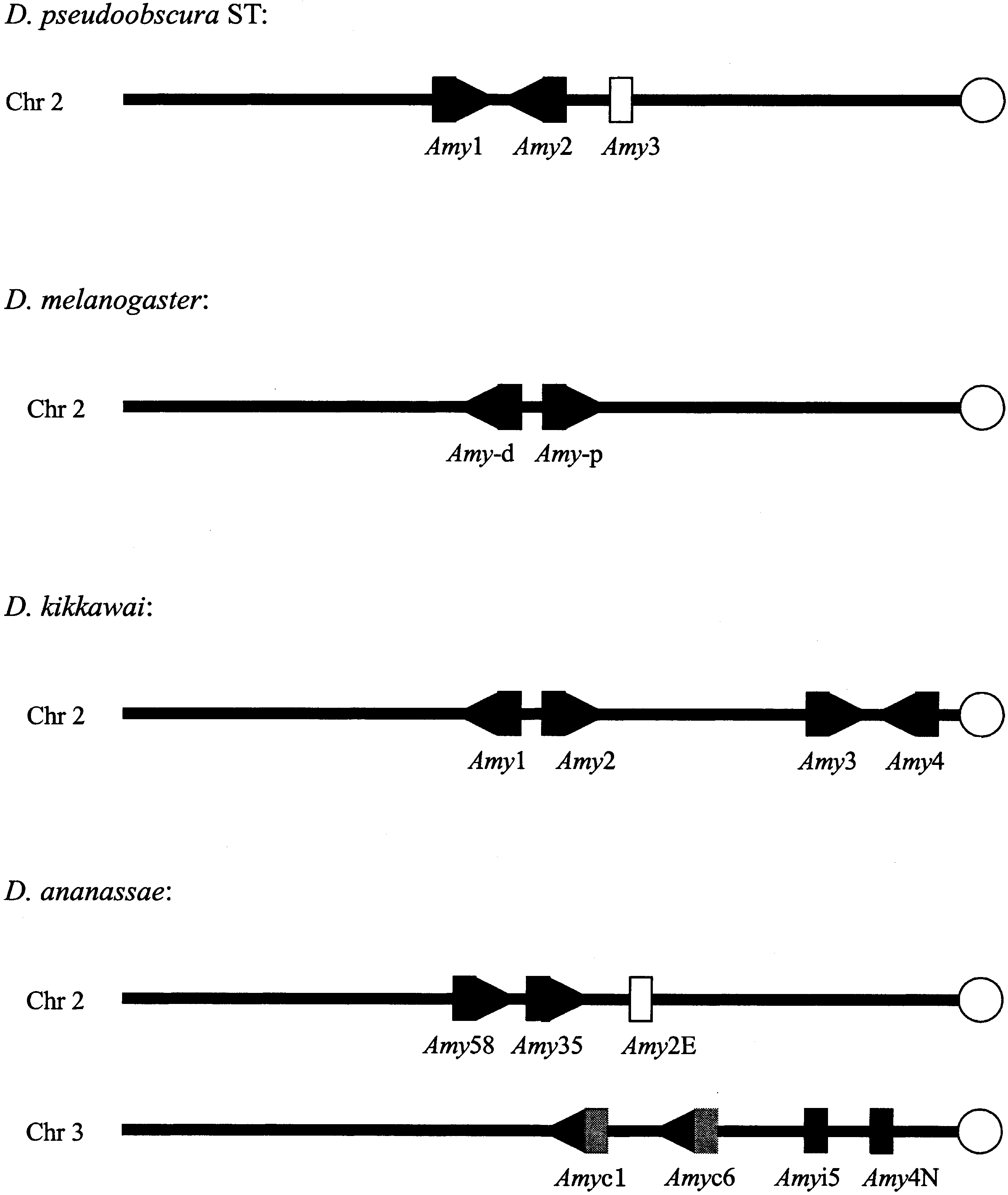 gene clusters in D. melanogaster,D. pseudoobscura ST, D. kikkawai, andD. ananassae. Open circles refer tocentromeres of chromosomes. Orienta-tions of the Amy genes are indicated byarrows if they are known. Open rec-tangles indicate a pseudogene or partialsequence available. Genes in gray donot have significant expression infor-mation available or are Amyrel genes.
gene clusters in D. melanogaster,D. pseudoobscura ST, D. kikkawai, andD. ananassae. Open circles refer tocentromeres of chromosomes. Orienta-tions of the Amy genes are indicated byarrows if they are known. Open rec-tangles indicate a pseudogene or partialsequence available. Genes in gray donot have significant expression infor-mation available or are Amyrel genes.