Tadalafil zeichnet sich durch eine außergewöhnlich lange Halbwertszeit im Vergleich zu anderen PDE5-Inhibitoren aus. Diese pharmakokinetische Eigenschaft führt zu einer verlängerten Exposition des Wirkstoffs im Organismus. Die Eliminationsrate hängt von der hepatischen Aktivität des CYP3A4-Enzyms ab. Lipophile Eigenschaften unterstützen eine weite Verteilung in unterschiedlichen Geweben. Eine ausgeprägte Stabilität gegenüber Nahrungsaufnahme macht den Stoff besonders konstant in seiner Wirkung. Unter generischen Präparaten wird cialis online häufig mit einem vergleichbaren pharmakologischen Profil beschrieben.
Pii: s1471-4914(01)02017-2
TRENDS in Molecular Medicine Vol.7 No.8 August 2001
unlikely that neuronal loss in neurodegenerative
How do neurons die
diseases is solely accomplished by apoptosis. Anyproposed mechanisms of neuronal death shouldexplain this extraordinarily slow time course. in neurodegenerative Morphological and molecular hallmarks of individual neurodegenerative diseases diseases?
Each disease has its own hallmarks. These hallmarksare the obvious choice as parameters for the study ofspecific neural death. Ichiro Kanazawa
Senile plaques and neurofibrillary tangles in corticalneurons in AD
Given that neurons are post-mitotic cells, their life span is generally long
Extracellular senile plaques (SP) and intra-
enough to reach that of humans. However, sometimes neurons die without
neuronal neurofibrillary tangles (NFT) are cardinal
recognizable causes, as a result of a process called neurodegeneration. Apart
pathological hallmarks of AD. The main chemical
from when gene mutations can be correlated with disease, it is difficult to
component of the core of SP is amyloid β protein
pinpoint molecules that are responsible for neuronal death. Therefore,
[Aβ, a mixture of Aβ40 and Aβ42 proteins, which are
neurons living in a ‘sick state’ for many years might reveal important
produced by cleavage of an amyloid β protein
information about neuronal death. Systematic and extensive single-neuron
precursor (APP) by secretases]5. Presenilins, which
analysis of ‘sick’ neurons is expected to provide clues to the mechanisms of
have recently been identified as γ-secretases, are
neurodegeneration. Moreover, the elimination of putative triggering and
mutated in a subset of early-onset familial AD
promoting factors involved in neurodegenerative disease might prevent
(Refs 6,7). Given that Aβ added to cultured neurons
disease progression.
is toxic to the cells8, neuronal death is expected tooccur by intracellular accumulation of Aβ. The
Neurodegenerative disorders are characterized
second hallmark of AD is the intracellularly
clinically by insidious onset and slowly progressive
deposited NFT, which is a polymerized, argyrophilic
course, and are frequently hereditary. Pathologically,
abnormal structure composed of hyperphosphorylated
these diseases share a common feature: the selective
tau protein9. By analogy with studies of Down’s
loss of a particular subset of neurons for unknown
syndrome, AD pathology might begin with the
reasons [e.g. cerebral cortical neurons in ALZHEIMER’S
formation of SP and years later proceeds to the
DISEASE (AD) (see Glossary), substantia nigra neurons
formation of NFT (Ref. 10). In this respect, it is
in PARKINSON’S DISEASE (PD), spinal motoneurons in
worth noting that the tau-phosphorylating enzyme
AMYOTROPHIC LATERAL SCLEROSIS (ALS), and striatal
(tau phosphokinase, TPKI) could be one of the
small neurons in HUNTINGTON’S DISEASE (HD)].
missing links between SP and NFT. Indeed,
Apoptosis has recently been implicated as a possible
the activation of intracellular TPKI is induced by the
mechanism for neuronal death in neurodegenerative
extracellular Aβ (Ref. 11). Neuronal loss in the
diseases. However, there is no direct and convincing
superior temporal gyrus of AD patients exceeds
evidence of apoptosis in human brains, and the
the number of NFT-positive neurons by more than
mechanisms of neuronal death in neurodegenerative
sevenfold1. Therefore,the majority of neurons can
diseases are still unknown. Here, I will discuss
die without developing NFT. Thus, although Aβ in
several proposed mechanisms of neuronal death in
individual diseases. In addition, I will put forward
molecules for the understanding of the AD
the concept of a long-standing ‘sick state’ of
pathogenesis, there is still a gulf between hallmarks
remaining neurons and the possible underlying
mechanisms of neuronal ‘sickness’. Neuronal loss in neurodegenerative diseases is an
The cardinal pathological hallmark of PD is the
extremely slow process
appearance of hyaline-like intracytoplasmic
Each neurodegenerative disease has its own clinical
inclusions, Lewy bodies (LB). LB are found in the
course. AD, PD and HD, for example, begin
remaining dopaminergic neurons in the substantia
gradually and progress slowly for more than
nigra and other nuclei. Following the identification of
10–20 years. By contrast, ALS progresses rapidly
an α-synuclein gene mutation as the cause of
and the disease process usually lasts only 2–3 years.
dominantly inherited rare PD (Ref. 12), α-synuclein
Corresponding to the clinical course, the time course
has also been established as the major component of
Ichiro Kanazawa
of neuronal loss is slow in AD and PD, and relatively
LB in sporadic PD. Indeed, a mutation in the gene
rapid in ALS (Refs 1–3) (Fig. 1). The speed of
encoding α-synuclein leads to the loss of
neuronal loss in neurodegenerative diseases is much
dopaminergic neurons and intra-neuronal inclusions
slower than that of apoptotic neuronal loss in
in a Drosophila model of PD (Ref. 13). Normal
developing nervous systems4. It is, therefore,
α-synuclein is localized in the presynaptic terminals,
http://tmm.trends.com 1471-4914/01/$ – see front matter 2001 Elsevier Science Ltd. All rights reserved. PII: S1471-4914(01)02017-2
TRENDS in Molecular Medicine Vol.7 No.8 August 2001
Glossary Alzheimer’s disease (AD): Neurodegenerative disease that begins
with insidious memory loss in late life. Subsequently, language
and visiospacial impairments occur. Frequency in over 65 years is
approximately 3–5%. AD is predominantly sporadic, with <20%being autosomal-dominantly inherited.
Amyotrophic lateral sclerosis (ALS): Characterized by muscle
atrophy or weakness in hands, feet or tongue from age 20
onwards, progressing rapidly. Respiratory impairments can be
life threatening. Frequency is about 1–3 cases per 100 000. CAG repeat: Glutamine-encoding tandemly repeated codon found
in the human genome. Abnormal expansion of CAG repeats
(to >40) can result in disease-causing polyglutamine stretches. Huntington’s disease (HD): Initial symptoms include chorteic
involuntary movement or intellectual deterioration in the 30s or
40s, but the disease typically progresses slowly. The HD-causing
mutation is an expansion of a CAG repeat in the IT15 gene. Parkinson’s disease (PD): Neurodegenerative disease that begins
with insidious tremor at rest, or slowness in movement on one
side of the body in middle to old age. Frequency is about 100
cases per 100 000 population. More than 90% of the patients are
RNA editing: A mechanism of post-transcriptional modification. A
single nucleotide can be substituted in the mRNA sequence; for
example, CAG (Gln codon) is substituted to CGG (Arg codon). Fig. 1. Superimposed
binds to synaptic vesicles, and is transported by the
axonal flows. The mutation in the α-synuclein gene
renders the protein devoid of vesicle-binding activity
and promotes accumulation-forming β-sheet
transgenic mice containing the mutated SOD1
structure14. However, the precise role of this protein
gene18. Moreover, given that the familial type is a
in neurodegeneration is still unclear. Recently, the
rare subset of ALS, SOD might not have a role in
parkin gene was identified as being responsible for
an autosomal-recessive form of juvenile-onset PD.
Parkin is thought to be a substrate for ubiquitination
Intranuclear inclusions in striatal small neurons of HD
leading to protein degradation by the proteasomal
HD is caused by an expansion of CAG REPEATS located
complex15. Because LB are not formed in this
in the coding region of the IT15 gene, whose product
recessive PD syndrome, a possible correlation
is named huntingtin. Abnormal huntingtin,
between parkin protein and the pathogenesis of
therefore, has an unusually long glutamine tract19.
sporadic PD is uncertain. Furthermore, the normal
Intranuclear inclusion bodies were found in the
function of parkin is also yet to be clarified.
striatal neurons of transgenic mice expressing the
CAG repeat containing exon 1 of the huntingtin
Intracellular inclusions in spinal motoneurons in ALS
gene20. The inclusion bodies are also found in the
The Bunina body in spinal motoneurons is the most
HD striatum and cortex. Inclusion bodies are
well known intracellular inclusion body associated
aggregates of a truncated form of protein containing
with ALS, but is still not fully characterized. Other
a polyglutamine stretch, ubiquitin, glyceraldehyde-
inclusion bodies in motoneurons of ALS are, unlike
3-phosphate dehydrogenase (GAPDH) and many
LB in PD, not uniform and are described by various
other proteins. However, there does not seem to be a
names such as argyrophilic, hyaline, conglomerate
correlation between the formation of inclusion
and skein-like inclusions. Most of them are,
bodies and neuronal death in cultured neurons that
however, an accumulation of phosphorylated
express abnormal huntingtin21. By analogy with
neurofilaments16. These findings, and the presence of
other polyglutamine diseases, the incorporation of
axonal spheroids suggest that ALS might be strongly
related to the disturbance of neurofilament
stretch into the nucleus itself, rather than the
function. Recently, mutations in the intracellular
formation of aggregates, is now thought to cause
Cu2+–Zn2+-dependent superoxide dismutase (SOD1)
neuronal death through disturbing normal
gene were discovered in rare familial ALS (Ref. 17).
functions of transcription factors22. The actual
Spinal motoneurons from familial ALS patients
relationship between the disturbed gene expressions
frequently bear intracellular inclusions that are
and the consequent neuronal death is still a
immunoreactive for an antibody against SOD1.
matter of debate. In summary, hallmarks of
Because SOD1 acts as a detoxifier of free radicals, a
neurodegenerative diseases are valuable clues for
mechanism of neuronal death is expected to be
understanding the pathogenesis of the disease.
related to the loss of SOD activity. However, there is
However, it is important to recognize that disease
no positive correlation between the gene defect and
hallmarks are not necessarily parameters for
motoneuron loss, not only in patients but also in
TRENDS in Molecular Medicine Vol.7 No.8 August 2001
Molecular mechanisms of ‘sick state’ of neurons
Whatever the initial trigger of neuronal death inneurodegnerative diseases is, consequent events canproceed insidiously, gradually or episodically. Eachneuron has its own optimal intraneuronalbiochemical conditions such as intracellular pH,water content, concentrations of oxygen, glucose,ATP, second messengers and Ca2+ ions. In ‘sick’neurons, these conditions might deviate slightlyfrom the optimum, without exceeding the life-threatening limit for neurons. It is possible,therefore, that a long-standing unfavorable living
condition might make neurons consume their energyinsidiously and after many years lead to theneuronal death. Possible mechanisms of ‘sickness’ ofneurons are summarized in Fig. 3.
Aging processBecause aging proceeds insidiously from middle life (for a review see Ref. 29), similar toneurodegenerative diseases, one might hypothesizethat neurodegenerative diseases are caused by anaccelerated aging process. Indeed, a certain number
of neurons are lost with age30: nigral neurons are
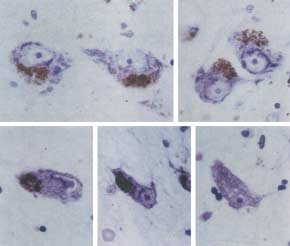
Fig. 2. Morphological ‘Sick neurons’
most severely affected, cortical neurons next and
In PD patients, less than 20% of nigral neurons
spinal motoneurons least. However, the speed of
remain 20 years after onset of the disease. Because
neuronal loss in AD and PD is notably faster than
they are destined to die, these remaining neurons
might provide important insight into neuronal
degeneration. Some of the remaining neurons show
‘degenerative changes’ in terms of size, shape and
significantly increased in the brains of elderly
morphology of neuronal soma and dendrites.
people. Nonenal might contribute to membrane
Although these changes are not specific, they might
damage and increased susceptibility to free radicals
represent definite signs of ‘sickness’ of neurons.
and consequently lead to serious disturbance of
For example, the density of dendritic branches of
neuronal structures and functions31, and 8-OHdG
most cortical neurons becomes coarse even in the
might be a marker of DNA oxidation. Moreover,
early stage of AD (Ref. 23). In addition, significantly
inactive enzymes, oxidized proteins and structurally
reduced numbers of dendritic spines and synaptic
altered proteins increase with age in the brain29. The
terminals were noticed in AD cortex24. These findings
amino acid groups of proteins non-enzymatically
support the decreased synaptic function in AD brain.
react with glucose or other monosaccharides, a form
In the substantia nigra of PD patients, the remaining
of post-translational modification. The products of
neurons show condensation of cytoplasm and nuclear
this reaction further produce, through oxidations or
indentation of neurons25. Quantitatively, 4–40% of
dehydrations, more complex protease-resistant
dopaminergic nigral neurons in PD were reported to
large molecules, for example, advanced glycation
show apoptotic cell fragmentation and autophagic
end-products (AGE), which increases with age. AGE
degeneration26. Indeed, our laboratory observed that
promotes inter- and intra-molecular crosslinking,
nearly 50% of remaining nigral neurons exhibit
and disturbs normal protein function32. It is
cellular shrinkage, cytoplasmic and nuclear
possible, however, that the aging process itself is not
condensation, and nuclear deformities (Fig. 2).
sufficient to kill neurons, but acts as a maintaining
In ALS, remaining spinal motoneurons shrink to
factor of the ‘sick state’ of neurons.
~70–80% in size27. By contrast, the remaining striatalneurons in dominantly inherited HD show little
shrinkage, but frequently show nuclear indentation28.
Oxidative stress (reviewed in Refs 33,34) is caused by
When taking the slow time course of neuron loss into
the enhanced production of harmful cellular oxidants:
consideration, surprisingly large numbers of neurons
free radicals (e.g. hydroxyl radical (.OH), superoxide
survive for more than 3–10 years in their seemingly
(O –), hydrogen peroxide (H O ), nitrogen oxide (NO)
atrophic and/or deformed state. These particular
and peroxynitrite (ONOO–)], or a failure of protective
‘sick’ neurons might be maintained at a lower level
mechanisms, including superoxide dismutase (SOD) or
than normal in terms of metabolism and function
glutathione peroxidase. Free radicals can enhance
membrane permeability to various molecules through
TRENDS in Molecular Medicine Vol.7 No.8 August 2001
Fig. 3. A hypothetical
peroxidation of membrane lipid, and lower the level of
neuronal activity. Of course, there are two protective
Glutamate is the most abundant excitatory
systems against free radicals in living cells,
neurotransmitter in the brain, and almost every
(1) enzymes converting radicals into harmless
neuron expresses glutamate receptors, either
compounds (e.g. SOD and glutathione peroxidase), and
permeating ions directly (AMPA/KA and/or NMDA
(2) non-enzymatic antioxidants (e.g. ascorbic acid or
types) or indirectly (metabotrophic type). An increase
tocopherol). If processes for free radical production are
of extracellular glutamate produces prolonged
somehow enhanced and protective processes reduced,
depolarization of neurons, inducing prolonged Ca2+
neurons could die. Indeed, there is evidence that free
influx into glutamate-receptive neurons, which then
radicals play a role in neuronal death not only in
leads to neuronal death (i.e. excitotoxicity )37,38. The
ischemic brain lesion but also of AD, PD, ALS and HD.
role for excitotoxicity in neuronal degeneration has
In PD, free radicals are easily produced with the help of
been extensively studied in ALS and HD. Although
Fe2+ in the course of the metabolism of dopamine.
the concentrations of glutamate in the spinal cord
Therefore, dopaminergic neurons are always exposed
and the brain of sporadic ALS patients are not
to free radicals. Evidence of a role of oxidative stress in
increased39, the predominant high-affinity
AD and HD is also accumulating35,36.
TRENDS in Molecular Medicine Vol.7 No.8 August 2001
specifically in astrocytes is lost in the ALS spinalcord. This might be a result of aberrant mRNA
caused by splicing errors40, causing an increase ofavailable glutamate in the peri-motoneuronal
environment. mRNA for the glutamate receptor 2(GluR2) subunit, which strongly regulates Ca2+conductance of the AMPA/KA receptor, is editednormally at the Gln/Arg residue in the subunitassembly. Recently, in the ventral horn of ALS
patients, GluR2 RNA EDITING was shown to besignificantly reduced41. This can lead to a continuousCa2+ influx through AMPA/KA receptors, therebymaking motoneurons vulnerable to various
endogenous or exogenous adverse insults. Indeed, ina mouse model of cerebellar ataxia, a causativemutation in the gene (GluRδ2) in the lurcher mouseleads to continuous Ca2+ influx and cerebellar
Reduced protein synthesisUsing DNA microarray techniques, a recent study
with a mouse model of HD revealed that in the earlystage of the disease, the brain expresses reducedlevels of mRNA of certain receptors and second
messengers, but not of mitochondrial proteins orapoptosis-related proteins43. Indeed, the dopamine-
Fig. 4. Ultramicro RT-PCR analysis of genes expressed in a single
synthesizing enzyme tyrosine hydroxylase (TH) and
human neuron. Brains were obtained at autopsy. Frozen 20µm sliceswere cut by cryostat-microtome. After a freeze-dry procedure, a single
its mRNA has been reported to be reduced in the
neuron was dissected from the substantia nigra using an excimer laser
remaining nigral neurons of PD (Ref. 44).
microdissector. PCR primers were designed to amplify the tyrosine
Preliminary single neuron analysis also showed that
hydroxylase (lane 1), dopa-decarboxylase (lane 2), α-synuclein (lane 3)
the remaining nigral ‘sick’ neurons in PD patients
and the ubiquitously expressed GAPDH (lane 4) genes. RT-PCR
products appeared as bands in a healthy control (C) and a Parkinson’s
definitely express GAPDH mRNA at normal levels
disease patient (PD, three different neurons; a–c). N indicates a
but express less than normal levels of TH, dopa
negative control without RNA templates (Jeong, S.M. et al.,
decarboxylase and α-synuclein mRNAs (Fig. 4).
unpublished) All four proteins examined are expressed in a nigral
These lines of evidence suggest that it is important to
‘sick’ neuron of PD patient (a), whereas dopa-decarboxylase is
extremely reduced in another (b). The third ‘sick’ neuron (c) only
know the overall expression profile of neurons in ‘sick
state’ to clarify the mechanism of ‘sickness’.
dysfunction in AD could be successfully reversed by
Therapeutic implications of ‘sick’ neurons
this treatment. Apart from the possible reversal of
Although the ‘sick state’ of neurons is generally
‘sickness’ of neurons, there are several lines of
regarded as irreversible, one could speculate that
therapeutic trials, either experimental or clinical, for
‘sickness’ of neurons could go back to the normal state,
neurodegenerative diseases. First, the fetal nigral
if triggering or promoting factors for neuronal
tissues were transplanted to the striatum of PD
damage were eliminated. This assumption is based on
patients and a part of dopaminergic function was
a recent report of HD transgenic mice model using a
improved. Second, a glial cell line-derived
tet/off system45. The system makes it possible to turn
neurotrophic factor (GDNF) introduced directly into
off the expression of a transgene with oral
substantia nigra or indirectly by vectors provided
administration of tetracycline analogs at any age
protection of nigral neuronal death and functional
after birth. The damage of the striatal neurons of this
recovery of the nigra in the experimental model of PD
particular mouse model is definitely reversed by
(Ref. 48). Third, a recent experimental study
inhibition of the continuous expression of the mutant
demonstrated that grafts derived from human fetal
HD-causing gene. If extrapolated to HD and other
striatal tissue can survive, develop, and are
polyglutamine diseases in humans, the ‘sick’ neurons
could go back to normal by inhibiting the expression
transplantation into a patient with HD (Ref. 49).
of the mutant gene. In addition, vaccinations with Aβ
Finally, using mesenchymal cell-derived factor(s),
peptide were reported to reduce amyloid deposition in
mammalian ES cells successfully differentiated into
a transgenic mouse model of AD (Ref. 46). Moreover,
neurons, particularly dopaminergic neurons50. This
the learning ability of Aβ-vaccinated mice was found
achievement opens the door towards a cure of PD, and
to be superior to that of non-immunized mice47.
hopefully this therapeutic principle can be applied to
Therefore, it is possible to speculate that the cognitive
the other neurodegenerative diseases.
TRENDS in Molecular Medicine Vol.7 No.8 August 2001
References
18 Gurney, M.E. et al. (1994) Motor neuron
34 Cohen, G. and Werner,P. (1994) Free radicals,
1 Gomez-Isla, T. et al. (1997) Neuronal loss
degeneration in mice that express a human Cu,Zn
oxidative stress, and neurodegeneration. In
correlates with but exceeds neurofibrillary
superoxide dismutase mutation. ScienceNeurodegenerative Diseases (Calne, D.B., ed.),
tangles in Alzheimer’s disease. Ann. Neurol.
19 Duyao, M. et al. (1993) Trinucleotide repeat
35 Markesbery, W.R. (1997) Oxidative stress
2 Fearnley, J.M. and Lees, A.J. (1991) Ageing and
length instability and age of onset in Huntington’s
hypothesis in Alzheimer’s disease. Free Rad. Biol.
Parkinson’s disease: substantia nigra regional
selectivity. Brain 114, 2283–2301
20 Davies, S.W. et al. (1997) Formation of neuronal
36 Browne, S. et al. (1999) Oxidative stress in
3 Schiffer, D. et al. (1991) Ubiquitin in motor neuron
intranuclear inclusions underlies the neurological
Huntington’s disease. Brain Pathol. 9, 147–163
disease: Study at the light and electron microscope.
dysfunction in mice transgenic for the HD
37 Choi, D.W. (1988) Glutamate neurotoxicity and
J. Neuropathol. Exp. Neurol. 50, 463–473
diseases of the nervous system. Neuron 1, 623–634
4 Majno, G. and Jois, I. (1995) Apoptosis, oncosis,
21 Sadou, F. et al. (1998) Huntingtin acts in the
38 Bergeron, C. (1995) Oxidative stress: its role in
and necrosis. An overview of cell death. Amer.
nucleus to induce apoptosis but death does not
the pathogenesis of amyotrophic lateral sclerosis.
correlate with the formation of intranuclear
J. Neurol. Sci. 129 (suppl) 81–84
5 Selkoe, D.J. (1998) The cell biology of α-amyloid
39 Rothstein, J. et al. (1990) Abnormal excitatory
protein and presenilin in Alzheimer’s disease.
22 Shimohata,T. et al. (2000) Expanded
amino acid metabolism in amyotrophic lateral
polyglutamine stretches interact with TAFII130,
sclerosis. Ann. Neurol. 28, 18–25
6 Esler, W.P. et al. (2000) Transition-state analogue
interfering with CREB-dependent transcription.
40 Lin, C.L.G. et al. (1998) Aberrant RNA processing
inhibitors of γ-secretase bind directly to
in a neurodegenerative disease: the cause for
presenilin-1. Nat. Cell Biol. 2, 428–434
23 Mehraein, P. et al. (1975) Quantitative study on
absent EAAT2, a glutamate transporter, in
7 Sherrington, R. et al. (1995) Cloning of a gene
dendrites and dendritic spines in Alzheimer’s
amyotrophic lateral sclerosis. Neuron 20, 589–602
bearing missense mutations in early-onset
disease and senile dementia. Adv. Neurol.
41 Takuma, H. et al. (1999) Reduction of GluR2 RNA
familial Alzheimer’s disease. Nature 375, 754–760
editing, a molecular change that increases calcium
8 Yankner, B.A. (1996) Mechanisms of neuronal
24 Davies, C.A. et al. (1987) A quantitative
influx through AMPA receptors, selective in the
degeneration in Alzheimer’s disease. Neuron
morphometric analysis of the neuronal and
spinal ventral gray of patients with amyotrophic
synaptic content of the frontal and temporal
lateral sclerosis. Ann. Neurol. 46, 806–815
9 Spillantini, M. et al. (1998) Tau protein pathology
cortex in patients with Alzheimer’s disease.
42 Zuo, J. et al. (1997) Neurodegeneration in Lurcher
in neurodegenerative diseases. Trends Neurosci.
mice caused by mutation in ∆ 2 glutamate
25 Klaue, R. (1940) Parkinsonsche Krankheit
receptor gene. Nature 388, 769–773
10 Mann, D.M. (1989) Cerebral amyloidosis, aging
(Paralysis agitans) und postencephalitischer
43 Luthi-Carter. R. et al. (2000) Decreased
and Alzheimer’s disease; a contribution from
expression of striatal signaling genes in a mouse
studies on Down’s syndrome. Neurobiol. Aging
klinischanatomischen Differentialdiagnose. Arch.
model of Huntington’s disease. Hum. Mol. Genet.Psychiat. Nervenkrankh. 111, 251–321
11 Takashima, A. et al. (1993) Tau protein kinase I is
26 Anglade, P. et al. (1997) Apoptosis and autophagy
44 Kastner, A. et al. (1993) Tyrosine hydroxylase
in nigral neurons of patients with Parkinson’s
protein and messenger RNA in the dopaminergic
neurotoxicity. Proc. Natl. Acad. Sci. U. S. A.
disease. Histol. Histopathol. 12, 25–31
nigral neurons of patients with Parkinson’s
27 Kiernan, J.A. and Hudson, A.J. (1993) Changes in
disease. Brain Res. 606, 341–345
12 Polymeropoulos. M.H. et al. (1997) Mutation in
shapes of surviving motor neurons in amyotrophic
45 Yamamoto, A. et al. (2000) Reversal of
the α-synuclein gene identified in families with
lateral sclerosis. Brain 116, 203–215
neuropathology and motor dysfunction in a
Parkinson’s disease. Science 276, 2045–2048
28 Roos, R.A. and Bots, G.T. (1983) Nuclear
conditional model of Huntington’s disease. Cell
membrane indentations in Huntington’s disease. Drosophila model of Parkinson’s disease. Nature
46 Schenk, D. et al. (1999) Immunization with amyloid-β
29 Finch, C.E. and Day, J.R. (1994) Molecular biology
attenuates Alzheimer’s disease-like pathology in the
14 Jensen, P.H. et al. (1998) Binding of α-synuclein
of aging in the nervous system: a synopsis of the
PDAPP mouse. Nature 400, 173–177
to brain vesicles is abolished by familial
levels of mechanisms. In Neurodegenerative
47 Morgan, D. et al. (2000) Aβ peptide vaccination
Parkinson’s disease mutation. J. Biol. Chem.Diseases (Calne, D.B., ed.), pp. 33–50, Harcourt
prevents memory loss in an animal model of
30 Brody, H. (1980) The nervous system and aging.
Alzheimer’s disease. Nature 408, 982–985
15 Shimada, H. et al. (2000) Familial parkinson
48 Björklund, A. and Lindvall, O. (2000) Parkinson
31 Montine, T.J. et al. (1996) 4-Hydroxy-2-nonenal is
disease gene therapy moves toward the clinic.
ubiquitin-protein ligase. Nat. Genet. 25, 302–305
cytotoxic and crosslinks cytoskeletal proteins in
16 Hirano, A. et al. (1984) Fine structural
P19 neuroglial cultures. Am. J. Pathol. 148, 89–93
49 Freeman, T.B. et al. (2000) Transplanted fetal
observations of neurofilamentous changes in
32 Munch, G. et al. (1997) Advanced glycation
striatum in Huntington’s disease: phenotypic
amyotrophic lateral sclerosis. J. Neuropathol.
endproducts in aging and Alzheimer’s disease.
development and lack of pathology. Proc. Natl.Acad. Sci. U. S. A. 97, 13877–13882
17 Deng, H.X. et al. (1993) Amyotrophic lateral
33 Beal, F. (2000) Energetics in the pathogenesis of
50 Kawasaki, H. et al. (2000) Induction of midbrain
sclerosis and structural defects in Cu–Zn
neurodegenerative diseases. Trends Neurosci.
dopaminergic neurons from ES cells by stromal
superoxide dismutase. Science 261, 1047–1051
cell-derived inducing activity. Neuron 28, 31–49
Letters to the Editor
Trends in Molecular Medicine: a forum for comment and debate
The Editor welcomes correspondence from readers of Trends in Molecular Medicine on any articles published. Please stateclearly whether or not you wish the letter to be considered for publication. If a letter is considered suitable for publication, itwill be sent to the author of the original article for their response; both the letter and reply will be published together.
Please note: submission does not guarantee publication.
NUPATHE REPORTS POSTIVE PHASE ONE RESULTS FOR NP101 TRANSDERMAL THERAPY FOR ACUTE MIGRAINE Results presented today at American Headache Society meeting in Chicago show that NuPathe’s SmartRelief™Transdermal Drug Delivery Technology effectively delivers therapeutic levels of sumatriptan over a sustained period Conshohocken, PA (June 8, 2007) – NuPathe Inc., a privately
Maternity Managing A Miscarriage This leaflet is available in other languages and formats, please contact the Patient Quality team on 0151 702 4160 This is a brief outline of the problem and is not intended to replace discussion withmedical or nursing staff. Managing a miscarriage Experiencing a miscarriage is very distressing and may have a great impact on you,your partner a
 TRENDS in Molecular Medicine Vol.7 No.8 August 2001
Molecular mechanisms of ‘sick state’ of neurons
TRENDS in Molecular Medicine Vol.7 No.8 August 2001
Molecular mechanisms of ‘sick state’ of neurons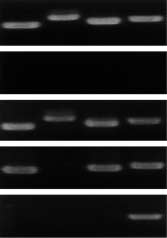 TRENDS in Molecular Medicine Vol.7 No.8 August 2001
specifically in astrocytes is lost in the ALS spinalcord. This might be a result of aberrant mRNA
caused by splicing errors40, causing an increase ofavailable glutamate in the peri-motoneuronal
environment. mRNA for the glutamate receptor 2(GluR2) subunit, which strongly regulates Ca2+conductance of the AMPA/KA receptor, is editednormally at the Gln/Arg residue in the subunitassembly. Recently, in the ventral horn of ALS
patients, GluR2 RNA EDITING was shown to besignificantly reduced41. This can lead to a continuousCa2+ influx through AMPA/KA receptors, therebymaking motoneurons vulnerable to various
endogenous or exogenous adverse insults. Indeed, ina mouse model of cerebellar ataxia, a causativemutation in the gene (GluRδ2) in the lurcher mouseleads to continuous Ca2+ influx and cerebellar
Reduced protein synthesisUsing DNA microarray techniques, a recent study
with a mouse model of HD revealed that in the earlystage of the disease, the brain expresses reducedlevels of mRNA of certain receptors and second
messengers, but not of mitochondrial proteins orapoptosis-related proteins43. Indeed, the dopamine-
Fig. 4. Ultramicro RT-PCR analysis of genes expressed in a single
TRENDS in Molecular Medicine Vol.7 No.8 August 2001
specifically in astrocytes is lost in the ALS spinalcord. This might be a result of aberrant mRNA
caused by splicing errors40, causing an increase ofavailable glutamate in the peri-motoneuronal
environment. mRNA for the glutamate receptor 2(GluR2) subunit, which strongly regulates Ca2+conductance of the AMPA/KA receptor, is editednormally at the Gln/Arg residue in the subunitassembly. Recently, in the ventral horn of ALS
patients, GluR2 RNA EDITING was shown to besignificantly reduced41. This can lead to a continuousCa2+ influx through AMPA/KA receptors, therebymaking motoneurons vulnerable to various
endogenous or exogenous adverse insults. Indeed, ina mouse model of cerebellar ataxia, a causativemutation in the gene (GluRδ2) in the lurcher mouseleads to continuous Ca2+ influx and cerebellar
Reduced protein synthesisUsing DNA microarray techniques, a recent study
with a mouse model of HD revealed that in the earlystage of the disease, the brain expresses reducedlevels of mRNA of certain receptors and second
messengers, but not of mitochondrial proteins orapoptosis-related proteins43. Indeed, the dopamine-
Fig. 4. Ultramicro RT-PCR analysis of genes expressed in a single