Tadalafil zeichnet sich durch eine außergewöhnlich lange Halbwertszeit im Vergleich zu anderen PDE5-Inhibitoren aus. Diese pharmakokinetische Eigenschaft führt zu einer verlängerten Exposition des Wirkstoffs im Organismus. Die Eliminationsrate hängt von der hepatischen Aktivität des CYP3A4-Enzyms ab. Lipophile Eigenschaften unterstützen eine weite Verteilung in unterschiedlichen Geweben. Eine ausgeprägte Stabilität gegenüber Nahrungsaufnahme macht den Stoff besonders konstant in seiner Wirkung. Unter generischen Präparaten wird cialis online häufig mit einem vergleichbaren pharmakologischen Profil beschrieben.
Pii: s0038-1098(00)00164-2
PERGAMON
Solid State Communications 115 (2000) 179–183
Poly(methyl acrylate-co-sodium methacrylate) ionomer
studied by solid state 13C T1r NMR
aDepartment of Physics, Jeonju University, Jeonju 560-759, South Korea
bDepartment of Polymer Science and Engineering, Chosun University, Kwangju 501-759, South Korea
Received 22 February 2000; accepted 6 April 2000 by A. Pinczuk
Abstract
The poly(methyl acrylate) (PMA) and poly(methyl acrylate-co-sodium methacrylate) containing 6.9 mol% of ionic groups
(PMANa-6.9) were studied by 13C CP/MAS NMR. The 13C spin–lattice relaxation times in a rotating frame, T1r, have beenmeasured as a function of temperature. Using these T1r spin–lattice relaxation times, we discuss the mobility, the correlationtime, and activation energy for the PMA and PMANa-6.9, respectively. The molecular motion in the PMANa-6.9 needs higheractivation energies than in PMA. It is worth noting that the motion of the 1-methyl carbons in the PMANa-6.9 ionomerdistinctly differs from that in the PMA homopolymer. The slow side of the T1r minimum associated with the 1-methyl carbonsis ascribed to stronger interactions between the polymer chains in the ionomer than in the homopolymer. ᭧ 2000 Published byElsevier Science Ltd. All rights reserved. Keywords: A. Polymers; B. Chemical synthesis; E. Nuclear resonances
1. Introduction
glassy polymer main chains in the low-to mid-kilohertzfrequency range, which are vital in determining mechanical
13C nuclear magnetic resonance (NMR) has proven to be
properties of polymers such as toughness [5]. The main
a very powerful technique for studying local dynamics of
chain motions of various polycarbonates at room tempera-
polymers, and 13C spin–lattice relaxation time is known to
ture have been determined from the 13C spin–lattice relaxa-
be an important experimental quantity for probing dynamic
tion measurements employing a high-resolution technique,
processes in polymers. Since the 13C nucleus is of low
including magic-angle spinning [6,7].
natural abundance, the relaxation is dominated by dipole
Polymers of low dielectric constants, which contain a
interactions with directly bonded hydrogens. By studying
relatively small amount of ionic groups, show the character-
the relaxation of the nuclei in different environment within
istic behavior of micro-separated materials [8,9]. This beha-
the chain, it is possible to obtain detailed pictures of chain
vior is known to be related to ionic aggregates within the
motions occurring in different parts of the chain. Thus, the
relatively nonpolar polymer matrix. The ionic aggregates,
measured relaxation data can be used to obtain information
called multiplets [10], induce the formation of a second
on the dynamic processes of polymer chains in different
phase, termed clusters [11]. These ionic polymers are
environment [1,2]. Recent studies have shown that more
known as ionomers and have both non-ionic and ionic
localized motions, i.e. mobility of the polymer backbone
nature. In order to explain the morphology and physical
chains, should be considered along with the conformational
properties of ionomers, Eisenberg, Hird and Moore
transitions in order to understand the differences in the
proposed a multiplet/cluster model in 1990 [11]. According
dynamics of the C–H vector at different sites of the back-
to the model, at very low ion contents, only multiplets are
bone [3,4]. The 13C T1r relaxation parameter is particularly
formed, and the material shows only one glass transition
informative since it is directly related to the motions of
(Tg), i.e. matrix Tg. The mobility of chains surrounding themultiplet is assumed to be restricted. As the ion content
increases, the regions of restricted mobility start to overlap. E-mail address: aeranlim@hanmail.net (A.R. Lim).
Once the dimension of overlapping regions of restricted
0038-1098/00/$ - see front matter ᭧ 2000 Published by Elsevier Science Ltd. All rights reserved. PII: S 0 0 3 8 - 1 0 9 8 ( 0 0 ) 0 0 1 6 4 - 2
A.R. Lim, J.-S. Kim / Solid State Communications 115 (2000) 179–183
polymer was filtered, and dried under vacuum at 70ЊC for1 day. To determine the acid content, the acid sample wasdissolved in a benzene/methanol (9/1 v/v) mixture andtitrated with standard methanolic NaOH to the phenolphtha-lein end point. The amount of acid groups was found to be6.9 mol%. To neutralize the acid polymer, the acid samplewas dissolved in a benzene/methanol mixture, and a prede-termined quantity of methanolic NaOH was added to givepoly(methyl acrylate-co-sodium methacrylate) (PMANa-6.9). The solution was freeze-dried and then dried undervacuum at 70ЊC for 1 day. The chemical structures of thepolymers are shown in Fig. 1. 2.2. 13C solid state NMR spectroscopy
The solid state NMR experiments were performed using a
Varian 300 NMR spectrometer at the Korea Basic ScienceInstitute in Taegu. The CP/MAS 13C NMR experimentswere performed at the Larmor frequency of 75.46 MHz. The samples of powder form were placed in a CP/MASprobe of 7 mm, and the magic angle spinning rate was5 kHz to minimize spinning sideband overlap. The p=2pulse time was 7.5 ms, corresponding to spin-locking fieldstrength of 50 kHz. 13C T
Fig. 1. Chemical structures of the polymers.
13C spin-locking pulse after a 0.8 ms CP preparation period.
The decay of the 13C magnetization in the spin-locking field
mobility, i.e. clusters, exceeds ca. 100 A
was followed for spin-locking time of up to 30 ms.
a second Tg, i.e. cluster Tg, attributed to the clusteredregions. As the ion content increases further, both the matrixand cluster Tgs of the ionomer increase. 3. Results and discussion
In this paper, we studied the structure of poly(methyl
The solid state structures and dynamics of PMA and
PMANa-6.9 were examined using the solid state NMR.
(PMANa-6.9) by 13C cross-polarization and magic angle
Fig. 2 shows the solid state 13C CP/MAS NMR spectrum
spinning (CP/MAS) NMR. The 13C spin–lattice relaxation
of the PMA. The spectrum consists of three signals at
time in a rotating frame, T1r, was also measured as a func-
chemical shifts of d 175:54; 52.31 and 41.89 ppm at
tion of temperature. From the results, the mobility of poly-
room temperature, which are assigned to carbonyl, 2-
mer chains, correlation time and activation energy for each
methyl ϩ b-methylene, and 1-methyl carbons, respectively.
carbon in the PMA and PMANa-6.9 were measured.
The carbon resonance peaks of 2-methyl and b-methyleneshow overlapping. Spinning sidebands are marked with anasterisk. The most intense signal is due to the 2-methyl and
2. Experimental
b-methylene carbons. In the case of the PMANa-6.9, the 13CCP/MAS NMR spectrum (not shown here) was similar to
that of the PMA. The chemical shifts of both the PMA andthe PMANa-6.9 were measured at various degrees of
The synthesis of poly(methyl acrylate) (PMA) homopo-
temperature, and were found to be nearly independent of
lymer and poly(methyl acrylate-co-sodium methacrylate)
The spin–lattice relaxation time in a rotating frame, T1r,
described elsewhere [12]. For convenience, only a brief
for each carbon of the two polymers was taken at different
summary of the procedure is given. The polymers were
degrees of temperature with variable spin-locks on the
prepared by solution polymerization of purified methyl
carbon channel following cross-polarization. The 13C
acrylate and methacrylic acid monomers using benzoyl
magnetization was generated by the cross-polarization
peroxide as the initiator at 60ЊC. Dried and distilled tetra-
after the spin-locking of protons. Then the proton Rf field
hydrofuran was used as the solvent. The conversion was less
was turned off for variable time t while the 13C Rf field
than ca. 10 %, and the polymer samples were recovered by
remained on. Finally, under high power proton decoupling,
precipitation into an excess of methanol. The precipitated
the 13C free induction decay (FID) was observed and
A.R. Lim, J.-S. Kim / Solid State Communications 115 (2000) 179–183
Fig. 2. Solid state 13C CP/MAS NMR spectrum of PMA at room temperature.
subsequently Fourier-transformed. The T1r values were
for each carbon in both the PMA and PMANa-6.9 poly-
obtained selectively by Fourier transformation of the FID,
mers as a function of temperature. In the case of the
following the end of spin-locking and repetition of the
PMA, the relaxation time of the 2-methyl, b-methylene,
experiment with variation of t. All the traces obtained in
and carbonyl carbons undergo motions on the slow side
the two polymers were fitted with the following single expo-
of the T1r minimum, while the 1-methyl carbons
undergo motions on the fast side of the T1r minimum. In the case of the PMANa-6.9 ionomer, the relaxation
Z t M0 exp Ϫt=T1r
time of all carbons undergo motions on the slow side of
Z and M0 represent the loss of magnetization and the
1r minimum. It is also seen that the T1r values
total nuclear magnetization of 13C in thermal equilibrium,
corresponding to the three peaks in the PMANa-6.9
are longer than those in the PMA. The 2-methyl, b-
For the studies of molecular motion in the experimen-
methylene, and carbonyl carbons also have longer
tal relaxation time, it is important to know whether the
relaxation time than the 1-methyl carbons. The relaxa-
relaxation time is located on the slow side of the mini-
mum or on the fast side of the minimum as a function
carbons similarly decreases with increasing temperature
of inverse temperature. At this point, it should be
for both the PMA and the PMANa-6.9 polymers. The
mentioned that the slow side of the curve can be inter-
most significant difference between the PMA and the
preted in such a way that the decrease in a T
PMANa-6.9 samples appears in the relaxation time of
indicates increased molecular motion; the fast side of
the 1-methyl carbons: it is in the fast motion in the
the curve can be interpreted in such a way that the
PMA case, while it is in the slow motion in the
PMANa-6.9 case. At this point, it should be mentioned
motion [15]. Fig. 3(a) and (b) shows the T
that the matrix glass transition temperatures, observed
A.R. Lim, J.-S. Kim / Solid State Communications 115 (2000) 179–183
mechanically dynamic, of the PMA and PMANa-6.9polymers are ca. 23 and 51ЊC, respectively. Thismeans that the polymer chains in the matrix phase inthe PMANa-6.9 ionomer experience more restrictedmobility than those in the PMA homopolymer. Thus,it can be expected that the chains in the ionomerwould show slower motion than those in non-ionic poly-mer, and that is what we observed here.
The T1r values can be related to corresponding values of
rotational correlation time, tC [15]. The rotational correla-tion time is the length of time that a molecule remains in agiven state before the molecule reorients. As such, the tC isa direct measure of the rate of motion. For the spin–latticerelaxation time in a rotating frame, the experimental valueof T1r can be expressed in terms of isotropic correlation time
tC as a very rough model for the molecular motions by thefollowing function [16,17]:
Here, gC and gH are the gyromagnetic ratios for the 13C
and 1H nuclei, respectively, N the number of directlybounded atoms, r the internuclear distance, ប h=2pwhere h is the Planck’s constant, vC and v H are the Larmorfrequencies of 13C and 1H, respectively, and v
50 × 2p × 103 rad=s is the spin-lock field. The types ofmotion and values of tC calculated from the Eq. (2) arelisted in Table 1. From the table, it is seen that the tC valuesof the 2-methyl and b-methylene carbons are much higherthan that of the 1-methyl carbons. Also clearly shown is thatthe 1-methyl carbons in the PMA undergo a fast motion asthe correlation time of 1:33 × 10Ϫ8 s whereas those in thePMANa-6.9 undergo a slow motion as the correlation timeof 1:93 × 10Ϫ4 s:
The temperature dependence of the relaxation time is a
Fig. 3. (a) Temperature dependence of the 13C spin–lattice relaxa-
tion time, T1r, for the PMA. (b) Temperature dependence of the 13Cspin–lattice relaxation time, T1r, for the PMANa-6.9.
where Ea is the activation energy for the molecular motion,R is the molar gas constant, and T is the absolute tempera-ture. Thus, a plot of the natural logarithm of relaxation timeas a function of inverse temperature is linear with a slopethat is proportional to the activation energy for motion. A.R. Lim, J.-S. Kim / Solid State Communications 115 (2000) 179–183
Table 1Activation energies (Ea), types of motion and rotational correlation time (tC) for the PMA and PMANa-6.9 polymers
Listed in Table 1 are also the activation energies for all the
Acknowledgements
carbons derived from the slope of the straight lines passingthrough the T1r data in the plot. From a comparison between
A.R. Lim thanks the Korea Basic Science Institute for the
those activation energies, the trends become clear. The acti-
support, and J.-S. Kim gratefully acknowledges the inter-
vation energies for all the carbons in the PMANa-6.9 are
disciplinary Research Program Grant 1999-2-308-002-3.
higher than those measured in the PMA. As was mentionedbefore, the molecular motion of the chains in the PMANa-6.9 clustered ionomer is restricted by the presence of ionic
References
groups, i.e. multiplets. Thus, when polymer chains gothrough a glass transition, the ionomer PMANa-6.9 needs
[1] F. Hearley, Prog. Nucl. Magn. Reson. Spectrosc. 13 (1979) 47.
a higher activation energy than the PMA homopolymer
[2] F. Hearley, Annu. Rep. NMR Spectrosc. 17 (1986) 179.
does. At this point, it is worth recalling that in dynamic
[3] D.J. Gisser, S. Glowinkowski, M.D. Ediger, Macromolecules
mechanical thermal analysis (DMTA) the activation ener-
gies for the polymer chains at the matrix glass transition
[4] S. Ravindranathan, D.N. Sathyanarayana, Macromolecules 28
were found to be ca. 570 and 480 kJ/mol for the PMANa-
6.9 and PMA, respectively [12]. This result is in good agree-
[5] J. Schaefer, E.O. Stejskal, R. Buchdahl, Macromolecules 10
ment with the activation energies obtained in the present
study, except that the activation energies obtained from
[6] J. Schaefer, R.A. Mckay, E.O. Stejskal, W.T. Dixon, J. Magn.
the DMTA experiments were ca. 50–60 times higher than
those obtained from the NMR experiments. However,
[7] T.R. Steger, J. Schaefer, E.O. Stejskal, R.A. Mckay, Macro-
considering that the DMTA and the NMR techniques
[8] A. Eisenberg, M. King, Ion-containing Polymers: Physical
measure activation energies of different sources, i.e. the
Properties and Structure, Academic Press, New York, 1977.
former from a large segment movement while the latter
[9] A. Eisenberg, J.-S. Kim, Introduction to Ionomers, Wiley,
from a nucleus movement, the difference in the magnitude
of activation energies obtained from the two techniques can
[10] A. Eisenberg, Macromolecules 3 (1970) 147.
[11] A. Eisenberg, B. Hird, R.B. Moore, Macromolecules 23
[12] J.-S. Kim, Y.H. Nah, S.-S. Jarng, W. Kim, Y. Lee, Y.-W. Kim,
4. Conclusions
[13] L. Laupretre, L. Monnerie, J. Virlet, Macromolecules 17
In the present study, the activation energies for each
carbon were obtained from the relaxation time as a function
[14] A. Spyros, D. Dais, R.H. Marchessault, J. Polym. Sci. Polym.
of temperature. It was found that the molecular motion in the
PMANa-6.9 needs higher activation energies than that in the
[15] M. Guo, Macromolecules 30 (1997) 1234.
PMA. Higher activation energies indicate higher rigidity of
[16] N. Bloembergen, E.M. Purcell, R.V. Pound, Phys. Rev. 73
the polymer chains in the PMANa-6.9 sample than in the
[17] G.P. Jones, Phys. Rev. 148 (1966) 332.
PMA sample. It is worth noting that the motion of the 1-
[18] J.L. Koenig, Spectroscopy of Polymers, ACS Professional
methyl carbons in the PMANa-6.9 ionomer distinctly differs
Reference Book, American Chemical Society, Washington
from that in the PMA homopolymer. The slow side of the
T1r minimum associated with the 1-methyl carbons is
[19] V.J. Mcbrierty, K.J. Packer, Nuclear Magnetic Resonance in
ascribed to stronger interactions between the polymer chains
Solid Polymers, Cambridge University Press, Cambridge,
in the ionomer than in the homopolymer.
Gender Differences in Sleep, Fatigue, and Daytime Activity ina Pediatric Oncology Sample Receiving DexamethasoneStacy D. Sanford, PHD, James O. Okuma, MS, Jianmin Pan, PHD, Deo Kumar Srivastava, PHD,Nancy West, BSN, Lynne Farr, PHD and Pamela S. Hinds, PHD, FAANSt Jude Children’s Research Hospital, MemphisObjective To examine gender differences in sleep, fatigue, and daytime activity in a samp
Informationen zur Neuen Grippe (Influenza A/H1N1) für Schulleitungen 1. Situationseinschätzung Deutschland Das Virus A/H1N1, das die Neue Grippe verursacht, kann leicht von Mensch zu Mensch übertragen werden. Das zeigt die ständig steigende Zahl der Erkrankten. Derzeit sind in Deutschland 16.835 Personen an der Neu-en Grippe erkrankt (Stand 04. September 2009). Die Mehrzahl d

 PERGAMON
PERGAMON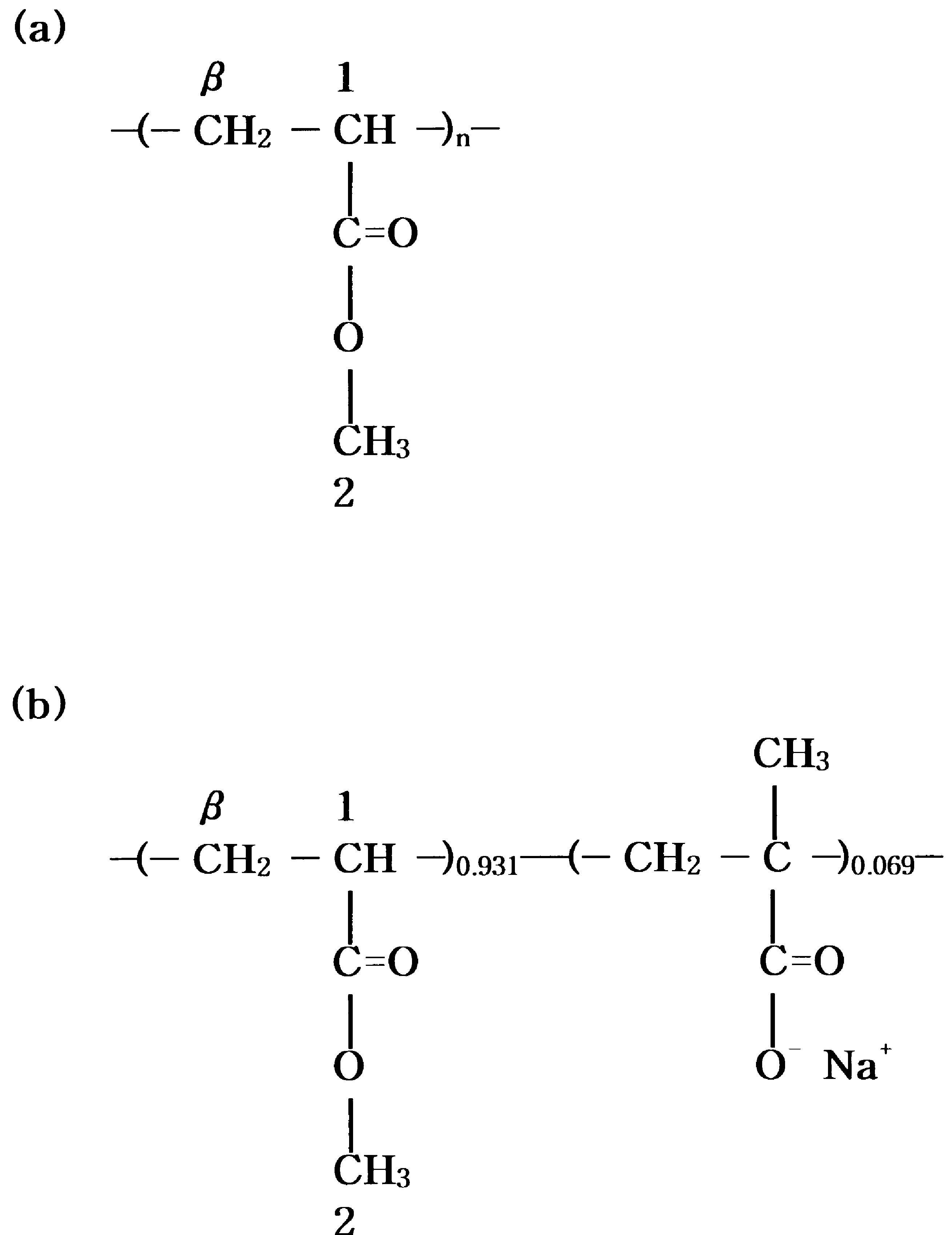 A.R. Lim, J.-S. Kim / Solid State Communications 115 (2000) 179–183
polymer was filtered, and dried under vacuum at 70ЊC for1 day. To determine the acid content, the acid sample wasdissolved in a benzene/methanol (9/1 v/v) mixture andtitrated with standard methanolic NaOH to the phenolphtha-lein end point. The amount of acid groups was found to be6.9 mol%. To neutralize the acid polymer, the acid samplewas dissolved in a benzene/methanol mixture, and a prede-termined quantity of methanolic NaOH was added to givepoly(methyl acrylate-co-sodium methacrylate) (PMANa-6.9). The solution was freeze-dried and then dried undervacuum at 70ЊC for 1 day. The chemical structures of thepolymers are shown in Fig. 1.
A.R. Lim, J.-S. Kim / Solid State Communications 115 (2000) 179–183
polymer was filtered, and dried under vacuum at 70ЊC for1 day. To determine the acid content, the acid sample wasdissolved in a benzene/methanol (9/1 v/v) mixture andtitrated with standard methanolic NaOH to the phenolphtha-lein end point. The amount of acid groups was found to be6.9 mol%. To neutralize the acid polymer, the acid samplewas dissolved in a benzene/methanol mixture, and a prede-termined quantity of methanolic NaOH was added to givepoly(methyl acrylate-co-sodium methacrylate) (PMANa-6.9). The solution was freeze-dried and then dried undervacuum at 70ЊC for 1 day. The chemical structures of thepolymers are shown in Fig. 1.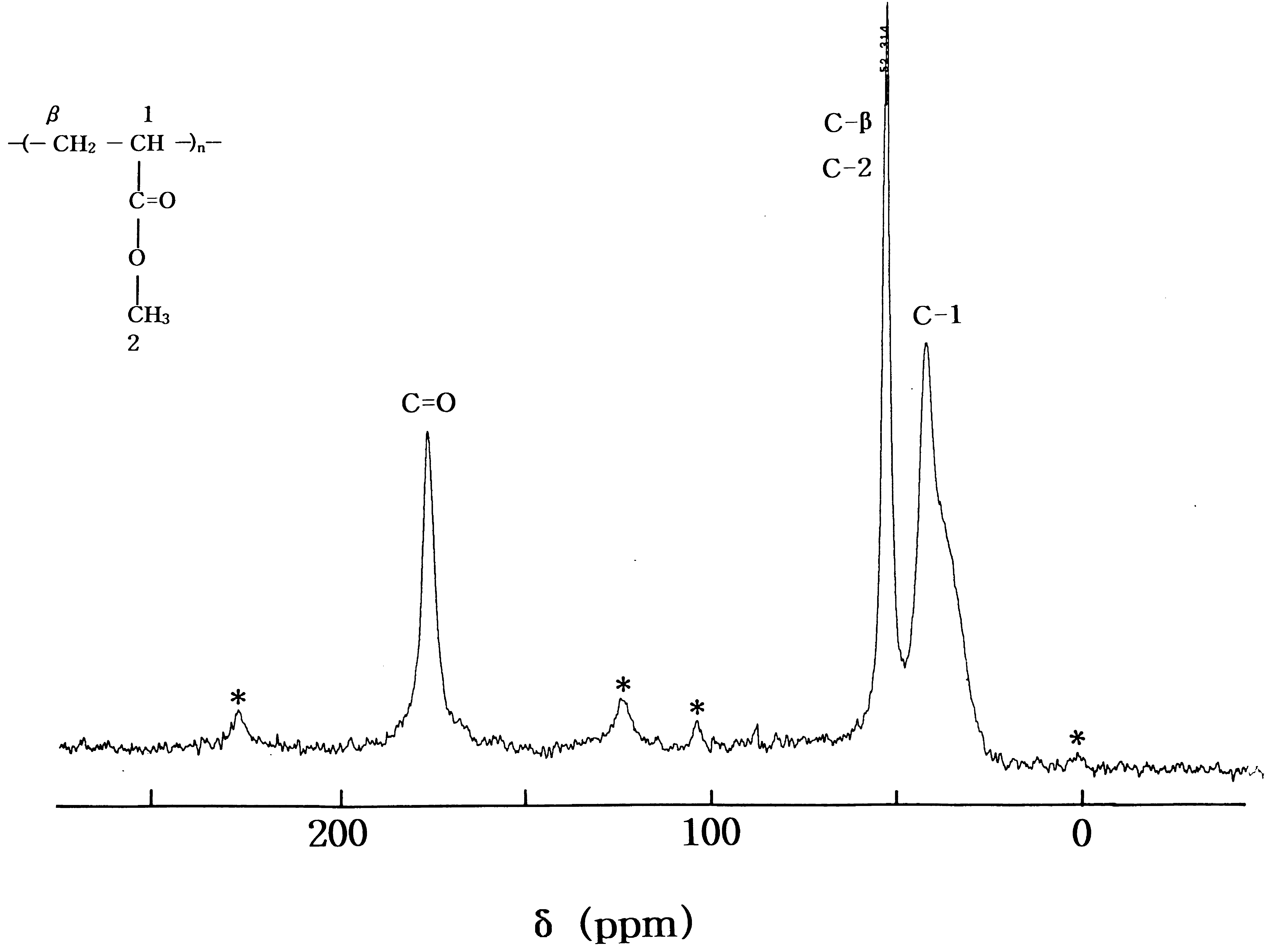 A.R. Lim, J.-S. Kim / Solid State Communications 115 (2000) 179–183
Fig. 2. Solid state 13C CP/MAS NMR spectrum of PMA at room temperature.
A.R. Lim, J.-S. Kim / Solid State Communications 115 (2000) 179–183
Fig. 2. Solid state 13C CP/MAS NMR spectrum of PMA at room temperature.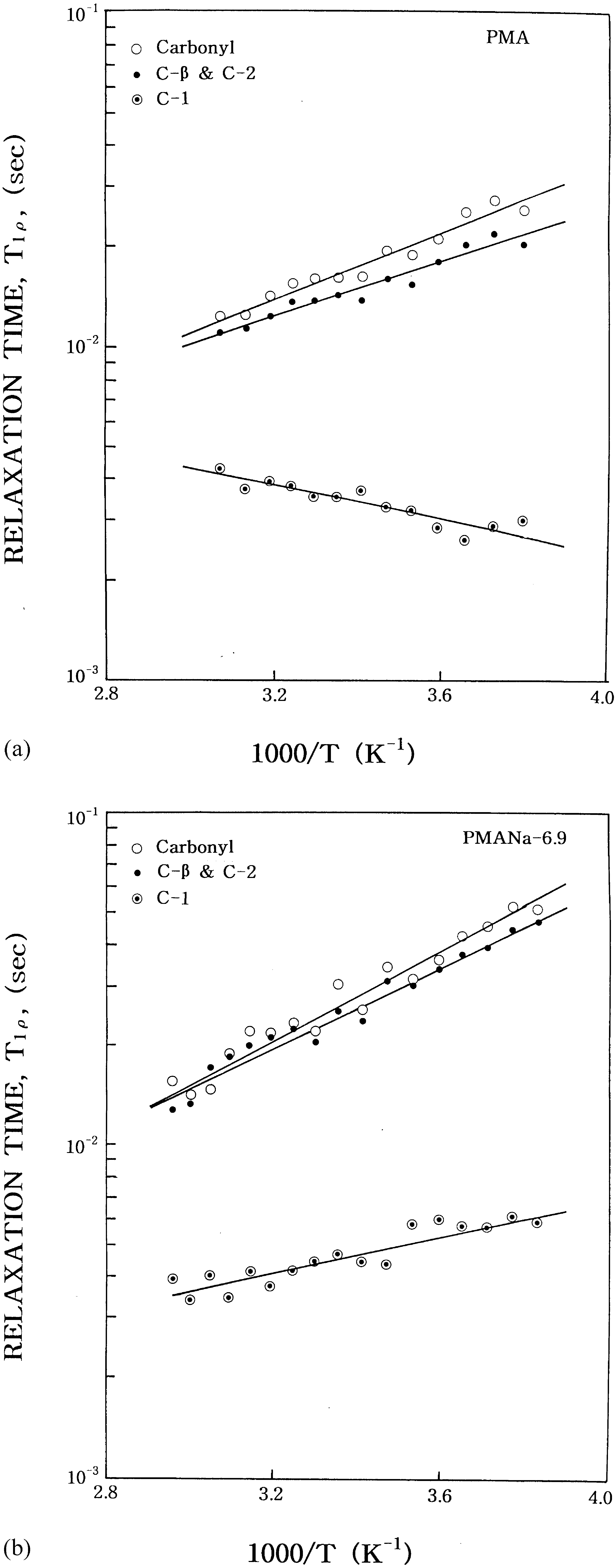 A.R. Lim, J.-S. Kim / Solid State Communications 115 (2000) 179–183
mechanically dynamic, of the PMA and PMANa-6.9polymers are ca. 23 and 51ЊC, respectively. Thismeans that the polymer chains in the matrix phase inthe PMANa-6.9 ionomer experience more restrictedmobility than those in the PMA homopolymer. Thus,it can be expected that the chains in the ionomerwould show slower motion than those in non-ionic poly-mer, and that is what we observed here.
A.R. Lim, J.-S. Kim / Solid State Communications 115 (2000) 179–183
mechanically dynamic, of the PMA and PMANa-6.9polymers are ca. 23 and 51ЊC, respectively. Thismeans that the polymer chains in the matrix phase inthe PMANa-6.9 ionomer experience more restrictedmobility than those in the PMA homopolymer. Thus,it can be expected that the chains in the ionomerwould show slower motion than those in non-ionic poly-mer, and that is what we observed here.