Tadalafil zeichnet sich durch eine außergewöhnlich lange Halbwertszeit im Vergleich zu anderen PDE5-Inhibitoren aus. Diese pharmakokinetische Eigenschaft führt zu einer verlängerten Exposition des Wirkstoffs im Organismus. Die Eliminationsrate hängt von der hepatischen Aktivität des CYP3A4-Enzyms ab. Lipophile Eigenschaften unterstützen eine weite Verteilung in unterschiedlichen Geweben. Eine ausgeprägte Stabilität gegenüber Nahrungsaufnahme macht den Stoff besonders konstant in seiner Wirkung. Unter generischen Präparaten wird cialis online häufig mit einem vergleichbaren pharmakologischen Profil beschrieben.
Caldwell.pdf
Compound optimization in early- and late-phase drug discovery: Acceptable pharmacokinetic properties utilizing combined physicochemical, in vitro and in vivo screens Gary W Caldwell Keywords β-adrenoceptor blockers, assays, bioavailability,
The RW Johnson Pharmaceutical Research Institute
Drug Discovery DepartmentWelsh and McKean RoadsSpring House
Introduction
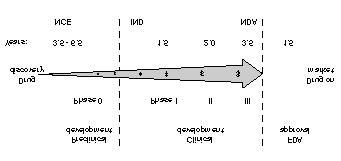
The drug discovery and development process is
scientifically complex and full of risk, and is therefore,expensive and time-consuming (Figure 1). Typically, a new
Current Opinion in Drug Discovery & Development 2000 3(1):30-41
chemical entity (NCE) is promoted from discovery into
preclinical development and if it succeeds in passing all
New chemical entities (NCEs) are abandoned in development
hurdles, it is submitted for an investigational new drug
primarily because of insufficient efficacy, safety issues and for
(IND) application and eventually enters phase I, II and III
economic reasons. Since efficacy and safety deficiencies are related
clinical development. If the compound passes all clinical
in part to pharmacokinetics (PK), uncovering PK defects as early
trials, it is submitted for a new drug application (NDA) and
in drug discovery as possible would be highly valuable in reducing
eventually enters the market-place. The average cost to
NCE failures in preclinical and clinical development. In this
discover and develop a NCE into a marketable drug in the
review, a strategy is put forth to integrate drug
USA, is typically hundreds of millions of dollars and
metabolism/pharmacokinetics and toxicology functions into drug
requires a decade or longer to reach the market-place [1]. It
discovery. Compound optimization in early- and late-phase drug
is clear that there is a critical need for pharmaceutical
discovery is covered, emphasizing physicochemical properties, in
companies to become more cost- and time-efficient in light
vitro absorption, metabolism and in vivo animal PK
of spiraling world healthcare expenditures. A significant
methodologies, primarily from the period 1998 to 1999. The
factor that governs the cost required for NCEs to become
present study also illustrates the idea of sorting oral bioavailability
marketed drugs is their high attrition rates in preclinical or
data into high/intermediate/low categories based on combining
clinical development [2]. The proportion of IND applications
high/low rank-ordered information from physicochemical
that fail has been estimated to be approximately 87% in
properties and in vitro absorption, metabolism and serum binding
phase I, 60% in phase II and 20% in phase III clinical studies
assays. It is shown that by combining the results from solubility,
[3•]. Coupling these attrition rates with the large
stability, absorption and metabolism assays, the
expenditures necessary for phase II and III clinical studies,
high/intermediate/low human oral bioavailability for a series of β-
produces the major financial problems associated with
blockers can be approximately predicted. This method has a high
pharmaceutical research. According to data compiled by
sample throughput and should be useful in rank-ordering the
DiMasi [2], 1943 INDs were filed in the USA between 1964
predicted oral bioavailability of large collections of compounds at
and 1989; the total number of IND applications that were
the lead optimization step of drug discovery. These results are
dropped before reaching NDA status was 1613 or 83%. In
useful for selecting compounds for future in vitro/in vivo
other words, during this 25-year period, approximately 1 in
correlation modeling or in vivo animal testing. This type of
6 NCEs nominated to IND status became a marketed drug. approach will improve the decision making process of compound
To make matters worse, only approximately 1 in 3 marketed
drugs typically generates sufficient income to recover the costs
Figure 1. The pharmaceutical discovery and development process. Compound optimization in early- and late-phase drug discovery Caldwell 31
associated with its discovery and development [3].
studies [5]. If defects in pharmacokinetic properties could
Therefore, it is generally true that the majority of
be recognized and corrected at the drug discovery stage
pharmaceutical discovery and development budgets and
before they entered development, more time and
time are spent on drug failures, and for NCEs that do go to
resources could be allocated to projects with real
market, only a few are genuinely profitable. Faced with this
potential. A compound optimization group would
low probability of success, the current trend of
certainly enhance any early clinical studies such as the
pharmaceutical companies merging is understandable.
drug selection interface group discussed above.
The abandonment of IND candidates is primarily
In this review, the importance of a compound
attributed to efficacy, safety and economic reasons. It has
optimization group that is broadly integrated within a
been reported for the period 1964 to 1989, that 46% of 1099
drug discovery department is discussed. Methods for
IND applications were discontinued due to unacceptable
compound optimization in early- and late-phase drug
efficacy (eg, weak or lack of efficacy), 27% due to safety
discovery are covered, emphasizing: (i) physicochemical
issues (eg, toxicity), 23% due to economics (eg, limited
properties (log P, pK , solubility, etc); (ii) in vitro and in
market) and 5% for miscellaneous reasons (eg,
vivo biophysical characteristics, such as absorption,
unclassified reasons) [2]. This trend is consistent with a
distribution, metabolism and excretion (ADME); (iii)
smaller, but more recent study by Kennedy [3], where 46%
pharmacokinetics; and (iv) toxicology. While the
of 121 NCEs in clinical development were discontinued
importance of human oral bioavailability data for
due to unacceptable efficacy, 40% due to safety issues, 7%
compound optimization in drug discovery is
due to economics and 7% for miscellaneous reasons.
unquestioned, it is rarely available due to cost- and time-
Therefore, insufficient pharmacological efficacy, human
consuming experimental challenges. The present study
adverse reactions and toxicity are estimated to account for
50% to 86% of the NCEs dropped from development. The
high/intermediate/low human oral bioavailability based
challenges of pharmacoeconomic predictions have
on rank-ordered physicochemical properties and in vitro
recently been addressed and will not be discussed here
[3]. Human in vivo efficacy and safety predictions of NCEs
are extremely difficult to forecast utilizing the presentpharmaceutical process, and the vast majority ofpharmaceutical companies are focused on reducing
Terminology and definitions
clinical development attrition rates by attempting to
Successful drug candidates typically have good biological
properties such as potency, selectivity, efficacy and oral
pharmacoeconomics at a much earlier stage than before.
bioavailability. It is important to have a reasonableunderstanding of these properties since some terms are
To address efficacy and safety attrition rates, some
used interchangeably in the literature, eg, potency for
companies are reorganizing their traditional discovery
efficacy and absorption for bioavailability. This section
and development departments from two independent
will briefly review their definitions. Potency refers to the
functions, to organizations that have a drug selection
amount of compound needed to produce a given
interface group between these departments. There are
biological effect and the terms activity and potency are
several jargon phraseologies given to this type of interface
used interchangeably in this review. Selectivity infers that
group, namely, 'proof-of-concept' or 'proof-of-principle'.
undesirable side effects are minimized or eliminated,
The primary mission of a drug selection interface group is
while efficacy refers to the maximum level of biological
to increase the quality and quantity of human data prior
effect a compound can produce. The oral bioavailability of
to full phase I clinical development. Typically, a drug
compounds is primarily dictated by the following serial
candidate is tested in a limited group of human subjects
rates: liberation, absorption, metabolism and elimination
(eg, 1 to 15) together with formal pharmacokinetic and
[6]. The simple diagram shown in Figure 2 can be used to
toxicology analysis. It is expected that this type of
define these terms. Oral ingestion is generally the safest,
information will indicate potential development problems,
most convenient and most economical method for
and thereby, significantly reduce the overall attrition rate
compound administration, and when a compound is
of NCEs in future clinical studies. Another strategy is to
given orally, the liberation rate is defined as the net
integrate drug metabolism/pharmacokinetics and
transfer of compound from the mouth and stomach to the
toxicology functions into a compound optimization group
small intestine. Typically, compounds are released from
either partially or entirely within drug discovery. It has
formulations that may depend upon disintegration,
been argued that uncovering human pharmacokinetic
dissolution, solubility, surface area and chemical or
defects (eg, oral bioavailability, half-life, metabolites,
enzymatic stability. In other words, the liberation rate
drug-plasma protein binding, etc) in drug discovery
represents the total amount of intact compound available
would be highly valuable in reducing NCE failures in
at the small intestine after oral dosing. The absorption rate
preclinical and clinical development, since efficacy and
involves the net transfer of compounds from the
safety deficiencies are related in part to pharmacokinetic
gastrointestinal fluid across primarily the small intestine
problems [4]. The jargon phraseology applied to this type
into the portal blood system. There are several
mechanisms of compound uptake; the main processes
pharmacokinetics' or 'just-in-time pharmacokinetics'. The
available are passive diffusion and carrier transport.
goal of this compound optimization group is to eliminate
Intestinal P-glycoprotein (P-gp) is another transporter
compounds with pharmacokinetic defects utilizing human
system that needs to be considered. P-gp is expressed at the
tissues, human-derived cell lines and/or in vivo animal
lumenal surface of the intestinal epithelium, and therefore,
32 Current Opinion in Drug Discovery & Development 2000 Vol 3 No 1 Figure 2.Routes from the gastrointestinal tract into the systemic circulation.
acts to oppose the uptake of compounds into the portal
This fraction or a percentage is normalized for the different
blood system [7]. The amount of compound that passes
compound doses (D) given by the two routes. Alternately, F
through the intestinal tissues must pass through the liver
and may be subjected to first-pass metabolism effects. Attimes, first-pass effects can prevent effective
concentrations of compounds from reaching the hepaticblood system and eventually the general systemic
Where f represents the net fraction of an oral dose liberated
circulation. The cytochrome P450 (CYP) enzyme system,
from the formulation that reaches the small intestine, the net
which is primarily located in the smooth endoplasmic
fraction absorbed across the apical membrane of the
reticulum of liver cells, and in smaller quantities in the
epithelial cell is denoted by f , and f represents the net
kidney, lung and gastrointestinal epithelium, is
fraction escaping the first-pass hepatic effect.
responsible for the monooxygenase metabolism ofcompounds [8]. Sometimes drug metabolites formed in
After introduction into the portal circulation system,
the liver are excreted back into the intestinal tract via the
compounds can bind to various constituents such as tissue
bile. These metabolites are either excreted in the feces or
proteins, cell proteins and blood proteins. Compound
reabsorbed into the portal blood system and ultimately
binding to various blood proteins and tissue proteins is
excreted in the urine. Compounds that reach the systemic
important because it can influence the therapeutic,
circulation either unchanged or as metabolites are
pharmacodynamic and toxicological action of certain
excreted by the urinary system. Sublingual and rectal
drugs. Competition for binding to tissues and blood
administration of compounds have the advantage that the
proteins is likely to occur between different compounds if
compound is somewhat protected from rapid first-pass
present at the same time. Human blood consists of three
metabolism by the liver, however, these routes are not as
major systems: (i) formed elements (ie, erythrocytes,
leukocytes and platelets); (ii) a fluid portion; and (iii) largeamounts of various salts [9]. The major cell body in the
The amount of orally-administered compound reaching the
blood is the erythrocyte (ie, red blood cell), which
systemic circulation is measured from the ratio of the area
comprises approximately 95% of the total cellular fraction
under the plasma-concentration versus time curve after oral
in the blood. There are three major components in the
administration (AUC) to that after intravenous (AUC)
erythrocyte capable of binding compounds - hemoglobin,
administration. Thus, the oral bioavailability (F) is defined
carbonic anhydrase and the cell membrane. If blood is
allowed to naturally coagulate, a clear straw-colored fluid(ie, serum) can be separated from the cellular fraction by
F = [(AUC)oral /( AUC)iv]*[ iv
centrifugation. In contrast, plasma is obtained by
Compound optimization in early- and late-phase drug discovery Caldwell 33
centrifugation of blood to which an anticoagulant was
relevant physicochemical, ADME, pharmacokinetics and
added immediately after removal from the body. Thus,
toxicology criteria in the decision making process of drug
serum contains water (90 to 92%), all blood proteins (6 to
discovery. In Figure 3, the organizational format of a
8%) and various salts (eg, 0.1 M NaCl), while plasma
broadly-based drug discovery department that is used to
contains water, proteins minus the clotting factors and
illustrate this idea is shown. Compound hit generation,
salts. The concentration of various serum proteins can
lead optimization and candidate selection steps are the
vary individually and on a daily basis by as much as 10%
three main decision points used to drive the overall
of their average value. It is also interesting to note that
process to produce drug development candidates. The
serum protein concentration levels can be highly affected
points have input from informatics (ie, bio- and
by certain physiological and pathological conditions.
chemoinformatics, molecular modeling, and computer
Human serum albumin (HSA), α -acid glycoprotein
and automation science), biology (ie, biochemistry,
(AGP), the high-density lipoproteins (HDL) and the low-
molecular biology, cell biology and pharmacology),
density lipoproteins (LDL), are the most important serum
chemistry (ie, combinatorial, parallel and scale-up
proteins responsible for the binding of compounds in
synthesis) and drug metabolism/toxicology (ie,
serum. Typically, HSA is largely responsible for serum
pharmacy, pharmacokinetics). This type of format
binding of acidic and neutral compounds, whereas AGP
naturally allows the composition of the department to
and lipoproteins bind mainly basic compounds. HSA and
change as the discovery process advances, ie, early drug
AGP serve as depots and transport proteins for numerous
discovery emphasizes more basic research in biology and
endogenous and exogenous compounds. Among the
chemistry, while late stage discovery shifts to more
endogenous substances bound with high affinity to blood
applied research. It allows for the elimination of
proteins are long chain fatty acids, L-tryptophan and
compounds utilizing a combination of activity and
bilirubin. The blood protein-exogenous complex (ie, blood
biophysical data at each decision point. This type of
protein-drug complex) acts as a transport mechanism to
organization also provides a maximum feedback loop
carry compounds to the sites of action; this transport is
extremely important for compounds that exhibit low
metabolism/toxicology scientists. In other words,
solubility in the water portion of the serum. In some cases,
structural modifications suggested by metabolic and
the circulating protein-compound complex also serves to
toxicity data are incorporated into the synthetic plan. We
replenish the free compound that is removed by various
will describe each decision point emphasizing the input
distribution and elimination processes. Thus, it maintains
from physicochemical, ADME and in vivo
free compound concentration at its therapeutic level and
pharmacokinetic assays for the selection of drug
provides a mechanism that prolongs the duration of
development candidates. It is not the intention of this
compound action. Therefore, determination of the
report to exhaustively review the array of important drug
concentration of unbound or bound compound with blood
metabolism/toxicology and absorption assays that
proteins is an important parameter to measure to establish
abound in the literature [11,12•,13-16], rather, it is to
the importance of oral bioavailability values.
highlight some important assays in the drug discoverysetting and make general comments concerning their use. The drug discovery process Traditionally, drug discovery groups have focused
The goal of the 'hit generation' step is to screen large
primarily on compound synthesis and target screening. In
compound libraries in a relatively short amount of time in
this type of environment, medicinal chemists synthesize
an attempt to find compounds that cause a specific
compounds in order to maximize in vitro or in vivo
biological response, ie, 'hits'. This step includes target
potency, selectivity and efficacy for a relevant biological
identification, selection, validation and high-throughput
target. Optimized structure-activity relationships (SARs)
screening (HTS) of large structurally-diverse compound
are generated based on in vitro potency versus structural
libraries. The productivity in target identification and
modifications; however, in many cases, all other
selection has improved with the development of
physicochemical properties (log P, pK , solubility, etc),
automated DNA sequencing, genomics databases and
ADME, pharmacokinetic and toxicological properties have
bioinformatic tools [17]. However, target validation
been ignored until a later time. As mentioned earlier,
remains a time-consuming process where assays are
uncovering pharmacokinetic defects is highly valuable in
typically performed without automation. HTS operations
reducing NCE failures since efficacy and safety
are highly automated to handle sample preparation, assay
deficiencies are related in part to pharmacokinetics [4].
procedures and large volume data processing. After each
The consequence of the exclusion of these properties in
step is optimized to operate efficiently, it is common to
drug discovery has been to waste time and resources in
screen 100,000 compounds in a 1- to 6-month period
development groups by putting them in a 'patch and
[18,19]. As new 'mix and read' detection assays are
mend' strategy. It is clear that the way to avoid this
developed, HTS is moving toward ultra-HTS which will
situation is to perform many of the traditional
screen over 100,000 compounds per day [20,21•].
development physicochemical, ADME, pharmacokinetics
Compound libraries are typically created by combinatorial
and toxicological studies in drug discovery [10•].
and parallel synthesis paradigms [22•], and HTS can
Unfortunately, many of these traditional in vitro and in
identify thousands of hits or only a few from these
vivo assays are not well adapted for the higher-throughput
libraries. The hit rate for HTS bioassays is set somewhat
screening that is necessary in drug discovery. Using a
arbitrarily, that is, the activity cut-off is lowered until an
hierarchical organizational approach and with recent
adequate number of hits is obtained. Cluster analyses can
methodological advances, it is feasible to add some
also be performed on thousands of hits in order to reduce
34 Current Opinion in Drug Discovery & Development 2000 Vol 3 No 1 Figure 3.The organizational format of a broadly-based drug discovery department.
the number of hits to an adequate number [23]. It is
approaches typically use QSAR techniques, knowledge-
interesting to note that libraries of either virtually
based systems or neural network modeling to predict
assembled compounds or actual compound libraries have
physicochemical properties and relevant biological
been computationally screened utilizing three- (3-D) or
parameters. While these calculations are well established,
four-dimensional (4-D) quantitative structure-activity
the accurate prediction of reliable data is still debatable.
relationship (3-D/4-D-QSAR) techniques to generate hits
There is a serious disadvantage in selecting or eliminating
[24,25]. Virtual screening is a promising new technique for
hits based on calculated or screening data of
the discovery of high-affinity hits [26]. Hits can also be
physicochemical, ADME, pharmacokinetics or toxicology
produced from 'me-too' compounds (close structural
data at this point. It should be remembered that it is rare
derivatives of a known active drug). The de novo design of
for a hit compound to have all of the desirable properties
new ligands bound in 3-D receptor protein structures
necessary for it to proceed directly to phase I clinical
using molecular modeling methods represents another
studies without structural modifications, furthermore, it is
source of hits [27]. The exact number of hits chosen is
generally true that any single property of a compound can
largely dictated by the in vitro potency, the compound
be optimized; however, this optimized property is often
patentability, the complexity of the analog chemistry,
achieved at the expense of other properties. If substantial
chemistry head count and the duration of the drug
structural manipulation to the prototype hit is required to
discovery program. In addition to these criteria, it could
obtain the desired potency, selectivity and efficacy, then
be of interest for certain projects to incorporate
all favorable physicochemical, ADME, pharmacokinetics
physicochemical, ADME, pharmacokinetics and
and toxicology properties obtained in the original hit may
toxicology selection criteria at this decision point.
be reduced or forfeited in the process. Therefore, it is
Unfortunately today, there are no in vitro or in vivo HTS
conceivable that nothing will be gained by screening for
methods capable of assaying 100K compound libraries in a
biophysical characteristics at this point in drug discovery.
It is more advantageous to screen the analogs for
pharmacokinetics and toxicology properties. DNA
favorable physicochemical, ADME, pharmacokinetics and
microarray technology or DNA chips is a promising
toxicology properties during the lead optimization step.
method used to monitor changes in expression at themRNA level [28]. While unproven at this time, it is
Once useful structural prototypes have been obtained from
conceivable that the DNA chip will discover pertinent
the hit generation step, analog construction around these
toxicodynamic markers [29] that can be used in reporter
templates utilizing traditional solution- and solid-phase
gene, branched-DNA or other HTS assays for early
chemistries are initiated so as to improve, primarily, in vitro
toxicology assessment [30]. There has been considerable
potency [22]. This process is referred to as the 'hit-to-lead
effort made in computer-aided physicochemical property
optimization step' or 'lead optimization step'. The types of
[31], solubility [32,33], intestinal uptake [33], permeability
structural modifications incorporated into the hit are usually
[34], metabolism [35] and toxicity [36] predictions. These
governed by in vitro potency data and by the wisdom of the
Compound optimization in early- and late-phase drug discovery Caldwell 35
medicinal chemist. If the hit has a 3-D X-ray crystallographic
pK [45], octanol-water partition coefficients [46],
compound-receptor protein structure, computational
thermodynamic solubility-pH profiles [47], and liposomal
chemistry (ie, molecular modeling) techniques are an
membrane-water partition coefficients [48] utilizing
important method for lead optimization [27]. Typically,
potentiometric techniques have been developed.
hundreds to thousands of analogs are required in the lead
Immobilized artificial membrane (IAM) chromatography
optimization step to select compounds worthy of
can be used to model membrane partitioning of
advancement to the candidate selection step and receive
compounds [49]. IAMs are solid-phase membrane
more extensive in vivo animal testing. The exact number of
mimetics that are prepared by covalently immobilizing
analogs synthesized per biological target is largely dictated
monolayers of cell membrane phospholipids to silica
by the potency, efficacy and selectivity deemed necessary
particles at high molecular surface densities, and are used
for the target, the personnel available, the complexity of the
as a chromatographic stationary-phase to mimic the lipid
analog chemistry and the duration of the drug discovery
environment of a fluid cell membrane [50]. The analyte
program. Since the total number of analogs for any
retention (capacity) factors on IAM chromatographic
particular target is synthesized over some time period, an
columns have been shown to predict drug permeability
iterative strategy can be used to organize the workload. For
across the blood-brain barrier [51]. An alternative to IAM
example, if 1000 analogs per target are synthesized over a 1-
has been suggested recently utilizing lysophospholipid
year period, then for 30 targets, roughly 2500 samples per
micellar electrokinetic chromatography [52]. The Caco-2
month need to be tested. There are only a few pertinent
(immortalized human colon adenocarcinoma) and the
physicochemical properties, ADME and toxicology assays
MDCK (Madin-Darby canine kidney) cell lines have been
that have medium sample throughput capable of handling
used as an in vitro model for assessing membrane
2500 samples per month. Turbidimetric methods for
permeability [53,54]. These cell lines differentiate on
measuring kinetic solubilities of compound have been
microporous filter membranes into columnar epithelium
estimated to handle 6000 samples per month [33].
and form tight cellular junctions. The utility of liquid
Permeation properties are related to transcellular compound
chromatography/mass spectrometric (LC/MS) methods
absorption. The use of artificial membranes in a 96-well
to measure the apparent permeability (P ) coefficients of
plate-based format to predict permeation properties of
compounds from a Caco-2 cell culture intestinal model
compounds has been estimated to handle 10,000 to 20,000
samples/month [37]. Hepatotoxicity studies, which give
electrophoresis/frontal analysis has been used to screen
information on the maximum compound concentration
drugs interacting with human serum and human serum
compatible with cell survival, can be performed using the 3-
proteins [56]. The rate of metabolism utilizing rat, monkey
(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
and human primary hepatocytes has been demonstrated
(MTT) colorimetric method [38]. The assay is based on the
using a sample handling system for 96-well plates directly
ability of mitochondrial enzymes in live cells to reduce a
coupled to a LC/MS, ranking compounds based on their
tetrazolium salt into a colored formazan dye. A 96-well
resistance towards metabolism [39]. The rationale behind
plate-based assay using a multi-well scanning
these experiments is that compounds that are resistant to
spectrophotometer has been described [39]. Recently, an
metabolism are more likely to exhibit low in vivo first-pass
HTS technique utilizing rat liver microsomes and a pulsed
effects. Reverse transcription-polymerase chain reaction
ultrafiltration mass spectrometer has been developed
(RT-PCR) assays have been developed to study the
[40•,41]. Pulsed ultrafiltration-electrospray mass
induction of human cytochromes P450 (1A1, 2A6, 2E1 and
spectrometry is an attractive method for in vitro formation
3A3/4) mRNA in cell culture systems [57]. The various
and mass spectrometric characterization of metabolites in an
assays outlined above can be used to simply rank-order
automated fashion. It was stated that the method had the
compounds or to select compounds based on a validated
potential for automated HTS of up to 60 samples (ie,
in vitro/in vivo correlation. The ranking of compounds
profiles) per hour. Fluorometric assays for assessing
based on these assays should be considered as a first step
cytochrome P450 drug-drug inhibition for the five principal
to reduce the number compounds for future in vitro/in vivo
drug-metabolizing enzymes, CYP1A2, CYP2C9, CYP2C19,
correlation modelingor in vivo testing. To be successful,
CYP2D6 and CYP3A4 have been developed in a 96-well
compounds with the desired activity and the fewest in
plate-based assay [42,43,44••]. The judicious application of
vitro biophysical defects or the best in vitro/in vivo
these techniques in rank-ordering compounds for more
correlations must be selected. Based on our experience,
extensive in vitro/in vivo correlation modelingor in vivo
the combination of a thermodynamic solubility assay [47],
animal testing may be useful; however, one should be
a Caco-2 permeability assay [55], a hepatocyte or
cautious since the data will contain many false-positive and
microsome assay to estimate hepatic extraction ratios [39],
false-negative results. Experience indicates that
a drug-blood protein binding assay [56] and a P450 drug-
physicochemical, ADME, pharmacokinetic and toxicology
drug inhibition assay [42], gives the most pertinent
information is only essential for the subset of analogs that
information. Valuable information is also gained from
demonstrate the correct in vitro potency range deemed
studying the profile of metabolites produced in rat and
necessary for the project to be viable. Using the example
human hepatocytes or microsomes. It should be noted that
above, for 2500 samples per month, we have found it
interpretation of results from all of the assays listed above
necessary to assay approximately 10% of these
could be hotly debated, since it will be necessary to
compounds or 250 compounds per month. If we only
eliminate compounds with activity at this point. In a later
assay those compounds that are considered to have the
section, it is shown how a combination of rank-order
correct activity, then there are many low-throughput in
screening assays can provide a high/intermediate/low
vitro assays at our disposal. Approaches for measuring
prediction of human oral bioavailability.
36 Current Opinion in Drug Discovery & Development 2000 Vol 3 No 1
Once the selection criteria have been defined in the lead
Intuitively, one would suspect that maximum oral
optimization step, compounds are promoted to the
bioavailability (eg, ≥ 90%) should occur when a compound
'candidate selection' step where the goal is to assay
has a high liberation rate (eg, the net transfer of
compounds utilizing primarily in vivo animal models.
compound from the mouth and stomach to the small
Using in vivo data obtained from animal species and
intestine), a high absorption rate (eg, net transfer of
allometric scaling, human pharmacokinetic parameters
compounds from the gastrointestinal fluid across
can be predicted, such as volume of distribution,
primarily the small intestine into the portal blood system)
clearance, half-life and oral bioavailability [58]. Note that
and low metabolism rate (eg, first-pass effect). Of course
at this stage, the function of the chemistry group has
the minimum concentration of compound in the systemic
shifted to scale-up chemistry (Figure 3) that is necessary,
system (eg, low oral bioavailability, ≤ 10%) should occur
since larger quantities of compounds are required for in
when the compound has a low liberation rate, a low
vivo studies. The number of candidates at this point is
absorption rate and a high metabolism rate. All other
totally arbitrary. For example, assume we have rank-
combinations of liberation, absorption and metabolism
ordered 250 compounds from one analog series, our
can be considered as having intermediate oral
experience indicates that we only need to assay
bioavailability values using the above arguments. Thus, if
approximately 10% of these compounds or 25 compounds
we could devise liberation, absorption and metabolism
per target. There are again several low-throughput in vivo
assays that ranked compounds in a high/low manner, it
assays at our disposal. An effective approach for screening
should be possible to predict into which oral
a large number of samples is to simultaneously measure
bioavailability range a compound would fall by utilizing
multiple analytes in a single analysis. In vivo cassette-
the scheme in Table 1. The liberation rate of a compound
dosing pharmacokinetic studies utilizing LC/MS have
can be approximated by examining the dissolution rate
been successful via the oral [59] and intravenous routes
and the compound's chemical stability. Since compounds
[60]. Alternatively, by using automated procedures to
are primarily solutions or suspensions in drug discovery,
handle plasma preparation, and rapid LC/MS techniques
the dissolution rate can be approximately assessed by the
to perform quantitation, we measure the oral
solubility of the compound assuming that the surface area
bioavailability of singly-dosed compounds in a rapid
of the compound is not a factor. Thus, the dissolution rate
manner. With this technique, many hundreds of
will not be a limitation for compounds having reasonable
compounds per year can be measured. Utilizing these fast
aqueous solubility (> 0.1 mg/ml) [33]. The chemical
quantitation techniques, it is possible to perform short-
stability of compounds at low gastric pH values can be
term in vivo toxicity experiments in order, for example, to
approximately assessed by stability studies at pH 2 over a
study exposure levels. Microdialysis sampling is a flexible
time period equivalent to the compound's mean residence
technique for the study of in vivo pharmacokinetics of the
time in the stomach (human ≈ 75 min). That is, we can
extracellular fluid in the brain [61]. In recent years,
measure the disappearance of compounds at pH 2 for 75
microdialysis with LC/MS detection has emerged as an
min. Thus, compounds with aqueous solubility > 0.1
important tool in biochemical research [62]. The various in
mg/ml and chemical stability > 50% (arbitrarily selected)
vivo assays outlined above can be used to identify potential
can be considered to have high liberation rates, while
pharmacokinetic defects, such as low oral bioavailability,
those with solubilities < 0.1 mg/ml and chemical stability
short half-life, active metabolites, etc. Based on in vivo and in
< 50% can be considered to have low liberation rates
vitro data, attempts can be made to correct these problems,
(Table 1). All other combinations of solubility and stability
and if successful, these compounds are promoted to drug
will be considered to have a low liberation rate. The Caco-
development candidacy and enter preclinical development.
2 cell line is used as an in vitro model to study drugtransport in the intestinal epithelium [55]. Compounds
Prediction of human oral bioavailability at the
that are completely absorbed in humans have
lead optimization step
permeability rates typically > 1.0 x 10-6 cm/s, compounds
Based on equation 2, if accurate human in vitro/in vivo
that are absorbed > 1%, but ≤ 100%, have permeability
correlation models to predict the f , f and f were
rates of 0.1 x 10-6 to 1.0 x 10-6 cm/s and compounds that are
available, human oral bioavailabilities (F) could be
absorbed < 1% have permeability rates ≤ 1.0 x 10-7 cm/s.
estimated. There are several good reviews outlining
Thus, compounds with permeability rates > 1.0 x 10-6 cm/s
methods to predict human oral bioavailability based on in
can be considered to have high absorption rates, while
vitro (human/animal)/in vivo (animal)metabolism data
those with < 1.0 x 10-6 cm/s can be considered to have low
[63,64]. The individual variation in oral bioavailability
absorption rates (Table 1). It should be emphasized, that
measurements can be very large due to genetic and
these permeability cut-offs are somewhat variable
environmental factors, and thus, cannot be accurately or
depending upon the experimental condition (eg, stirred
readily predicted. To take into account this natural
versus unstirred water layers) [55], however, the separation
variability, our strategy is simply to sort oral
of the Caco-2 data into high/low categories is typically not a
bioavailability data into high/intermediate/low categories
problem. Once the compound has passed from the small
based on combining rank-ordered information from several
intestine to the portal system, the liver may metabolize a
in vitro assays. This method has a higher sample throughput
considerable portion of the compound prior to entering the
than in vitro/in vivo correlation modeling, and therefore,
systemic circulation. The prediction of metabolism rates can
should be useful in sorting large collections of compounds at
be studied using isolated human hepatocyte cells and/or
the lead optimization step for future in vitro/in vivo
various liver subcellular fractions [39,65]. The rate of first-
correlation modelingor in vivo animal testing.
pass metabolism can be roughly estimated into high/low
Compound optimization in early- and late-phase drug discovery Caldwell 37 Table 1.Oral bioavailability classified into high/intermediate/low categories based on in vitro rank-ordered liberation, absorption and metabolism data. Liberation ranking Absorption ranking Metabolism ranking Oral bioavailability
categories by examining the rate of drug disappearance (or the
vivo first-pass effect data, we observe that the in vitro human
rate of appearance of the metabolites) from human hepatocyte
hepatocyte cell data could approximately differentiate the in
or microsomal suspensions. Thus, compounds with hepatocyte
vivo first-pass effect data in a high/low manner. That is,
stability > 50% (arbitrarily selected) can be considered to have
propranolol, alprenolol and oxprenolol were highly
low metabolism rates, while those with hepatocyte stability <
metabolized in vitro and also exhibited a high in vivo first-pass
50% can be considered to have high metabolism rates (Table 1).
effect. Timolol (3), pindolol, acebutolol and atenolol were
Thus, a simple high/low strategy could be used to roughly
resistant towards metabolism by human hepatocytes and also
estimate first-pass metabolism effects. Alternately, when
exhibited a low in vivo first-pass effect. However, it should be
concentration studies and scaling techniques are performed, it
noted that metoprolol (6) did not fit this pattern, suggesting in
has been found that drugs that had high in vivo extraction
vitro human hepatocyte cell data obtained at a single
ratios had in vitro hepatocyte clearance values > 30 µl/min/106
concentration and time point do not completely correlate with
cells, while drugs that had low in vivo extraction ratios had inin vivo first-pass effect data. Finally, if we combine the
vitro hepatocyte clearance values < 10 µl/min/106 cells [65].
high/low liberation, absorption and metabolism result fromTable 2 with the scheme outlined in Table 1, we can predict the
An homologous series of β-adrenoceptor blocking drugs (β-
high/intermediate/low oral bioavailability for the β-blockers.
blockers, Figure 4 and Table 2) were chosen as model
Note that timolol and pindolol were correctly chosen as having
compounds to illustrate the above scheme, since their
high bioavailability, however, metoprolol was predicted to
pharmacokinetic parameters display a wide variability [66,67].
have a high oral bioavailability, while the experimental data
These compounds have molecular weights that range from 248
suggests its value is closer to an intermediate value. Clearly,
Da (4, pindolol) to 336 Da (1, acebutolol), and dissociation
the in vitro methods discussed here are able to distinguish β-
constants (pK ≈ 9) which minimize the influence of these
blockers with favorable characteristics. We believe the above
parameters on solubility, stability, absorption and metabolism.
procedure will work reasonably well for other analog series
The pseudo-thermodynamic solubility was measured using a
and probably worse for structurally-diverse sets of
simple LC/MS technique. Briefly, the compound was
saturated at pH 7.4 (ionic strength = 0.15), shaken for 30 min,
As mentioned earlier, the prediction of drug-serum binding is
filtered through a nylon filter and assayed by LC/MS. The
an important factor to understand along with oral
stability was measured by preparing the sample at pH 2 and
bioavailability. It has been reported that capillary
measuring the disappearance over a 75 min period. Data for
electrophoresis/frontal analysis (CE/FA) can be used in an
the β-blockers are listed in Table 2 and indicate that these
automated manner to estimate the blood protein-drug binding
drugs are all highly liberated. For the Caco-2 transport in the
association constants [56]. The percentage of drug bound in
apical to basolateral direction, 50 µM of the β-blocker of
serum samples or artificial mixtures of serum proteins samples
interest was placed in the insert (apical side) [55]. The insert
(eg, HSA + AGP) can also be determined. Briefly, CE/FA is
was moved seven times over a 1 h period to wells containing
used to determine the free drug concentration in a drug-
fresh buffer. These well (basolateral) samples were spiked with
protein binding equilibrium. In CE/FA, sample volumes that
an internal standard and analyzed by LC/MS. The Papp
represent approximately 5 to 7% of the total volume of the
coefficients obtained from this experiment are listed in Table 2.
capillary are injected onto the capillary to result in a frontal
The results for the β-blockers indicated that acebutolol and
peak shape for the drug. Based on the frontal theory, the free
atenolol (2) had a low absorption rate, while all of the other
drug concentration is directly determined from the height of
drugs were highly absorbed. If we compare the P coefficients
the frontal peak. In the normal CE polarity mode, the basic β-
to the in vivo human absorption data of the β-blockers (Table
blockers elute first and are resolved from the negatively
2), we observe that the P coefficients correctly distinguish the
charged serum proteins. We have shown that the binding
poorly absorbed β-blockers (acebutolol and atenolol) from the
capacity of the β-blockers with human protein mixtures of
highly absorbed β-blockers. After known drug concentrations
AGP, HSA, HDL and LDL has the same high/low ranking
(5 µM) were incubated with hepatocytes for 6 h, LC/MS
order as that obtained with human serum samples. The results,
procedures were developed to determine the amount of drug
from this experiment indicated that propranolol had
which was metabolized (ie, % metabolized) [39]. These results
significantly higher binding to these proteins than the other β-
are listed in Table 3 and indicate that propranolol (8),
blockers. Therefore, timolol and pindolol have higher oral
alprenolol (7) and oxprenolol (5) were highly metabolized. If
bioavailabilities and lower serum binding profiles than
we compare the in vitro human hepatocyte cell data with the in
38 Current Opinion in Drug Discovery & Development 2000 Vol 3 No 1 Figure 4. β-adrenoceptor blocking agents. 1 acebutolol 2 atenolol 3 timolol 4 pindolol (Sandoz) 5 oxprenolol 6 metoprolol 7 alprenolol 8 propranolol (Wyeth-Ayerst) Table 2. In vitro and in vivo biophysical data for a series of β-adrenoceptor blocking drugs. Solubility1 Stability2 Liberation In vivo Absorption Hepatocytes5 In vivo Metabolism acebutolol atenolol pindolol oxprenolol metoprolol alprenolol propranolol
1The drugs were prepared at pH 7.4 (ionic strength = 0.15) in an aqueous solution, shaken for 30 min, filtered through a nylon filter andassayed by LC/MS. The units are mg/ml. 2The stability (% remaining) was measured by preparing the sample at pH 2 and measuring the disappearance over a 75 min period. Thedata are normalized to the starting concentration at time = 0. 3Caco-2 P coefficients (see reference [55] for experimental details). The data are normalized to propranolol (7.5 ± 1.2 x 10-5 cm/s).
4In vivo human absorption (%) (see references [66] and [67]). The data are normalized to propranolol (90%). The net fraction absorbedacross the apical membrane of the epithelial cell is denoted by f .
5In vitro human hepatocyte cell data (% metabolized in 6 h at 5 µM of drug). See reference [39] for experimental details. The data arenormalized to the starting concentration at time = 0 for propranolol. In vivo human first-pass effect (%) (see references [66] and [67]). Thedata are normalized to propranolol (60%). 6The notation f represents the net fraction escaping the first-pass hepatic metabolism effect. Compound optimization in early- and late-phase drug discovery Caldwell 39 Table 3. Comparison of the predicted and experimental oral bioavailability values for a series of β-adrenoceptor blocking drugs. Liberation Absorption Metabolism Predicted oral Experimental oral ranking1 ranking1 ranking1 bioavailability2 bioavailability3 acebutolol atenolol pindolol oxprenolol metoprolol alprenolol propranolol
1The ranking of the drugs was based on results from Table 2.
2The ranking of the oral bioavailability was based on the system used in Table 1. 3The in vivo human oral bioavailability (%) values are from references [66] and [67]. Conclusion References
This review has focused on integrating drugmetabolism/pharmacokinetics and toxicology functions into
drug discovery in order to reduce the attrition rates in clinical
development. Three main decision points are used in drug
1. DiMasi JA: Risks, regulation, and rewards in new drug
discovery to drive the overall process to produce superior
development in the United States. Regul Toxicol Pharmacol
drug development candidates. These include hit generation,
(1994) 19:228-235.
lead optimization and candidate selection steps. Theelimination of compounds occurs at each decision point
2. DiMasi JA: Success rates for new drugs entering clinical
utilizing a combination of activity, physicochemical properties,
testing in the United States. Clin Pharmacol Ther (1995)
absorption, metabolism, in vivo pharmacokinetics and toxicity
58:1-14.
assays. This type of organization provides a maximum
3. Kennedy T: Managing the drug discovery/development
feedback loop between medicinal chemists and drug
interface.Drug Disc Today (1997) 2:436-444.
metabolism/toxicology scientists. Literature has been cited
• A good review of the skill sets necessary to manage an interface
primarily between 1998 to 1999. At the lead optimization step,
between discovery and development.
a combination of a thermodynamic solubility assay [47], aCaco-2 permeability assay [55], a hepatocyte or microsome
4. Caldwell J: The importance of drug metabolism studies for
assay to estimate hepatic extraction ratios [39], a drug-blood
efficient drug discovery and development.Yakubutsu Dotai
protein binding assay [56], and a cytochrome P450 drug-drug
(1996) 11(1):119-126.
inhibition assay [42] gives the most pertinent information. The
5. Caldwell GW: Novel biophysical analytical methods: The
profile of metabolites produced in rat and human hepatocytes
utility of combining physicochemical and in vitro
or microsomes is also suggested at this step. These higher-
physiological proprieties of drug compounds for estimating
throughput assays can be rank-ordered to prioritize
in vivo oral bioavailability.Second International Symposium on
compounds for more extensive in vitro/in vivo correlation
Drug-Drug Interaction Potential. Baltimore, MD, USA (1997).
modelingor in vivo animal studies. A simple example was
6. Benet LZ, Mitchell JR, Sheiner LB: Goodman and Gilman's -
used to illustrate this idea. Utilizing a series of β-adrenoceptor
The Pharmacological Basis of Therapeutics. 8th Edition.
blocking drugs, a strategy was developed to sort oral
Gilman AG, Rall TW, Nies AS, Taylor P (Eds), Pergamon
bioavailability data into high/intermediate/low categories
based on combining rank-ordered information from several invitro assays. This oral bioavailability ranking procedure
7. Spahn-Langguth H, Baktir G, Radschuweit A, Okyar A, Terhaag
B, Ader P, Hanafy A, Langguth P: P-glycoprotein transporters
worked reasonably well for the β-adrenoceptor blocking
and the gastrointestinal tract: Evaluation of the potential in
drugs, and therefore, should work for other analog series. The
vivo relevance of in vitro data employing talinolol as model
predictions will probably be worse for structurally-diverse sets
compound.Int J Clin Pharmacol Ther (1998) 36:16-24.
of compounds. The candidate selection step is used to assaycompounds utilizing primarily in vivo animal models to gain
8. Lewis DF (Ed): Cytochromes P450: Structure, Function and
information on key pharmacokinetic parameters and drug
Mechanism. Taylor & Francis Ltd, London (1996):1-348.
exposure levels [60]. While the above procedure appears to be
9. Reif OW, Lausch R, Freitag R: High-performance capillary
a reasonable start, clearly much remains to be done to improve
electrophoresis of human serum and plasma proteins.Adv
drug selection in discovery and development. Chromatogr (1994) 34:1-56. Acknowledgements
10. Heykants J, Meuldermans W: Nonclinical kinetics and metabolism studies in support of the safety assessment of
The author would like to acknowledge the contribution of
drugs.Drug Inf J (1994) 28:163-172.
Per A Peterson, Michael Ernest, Keith Demarest, Joseph
• A good review of the studies necessary for preclinical and clinical
Gunnet, William Hageman, Eric Ericson, John A Masucci,
Patrick Sasso, William Jones, Amy Maden, MaryEvangelisto, Zhengyin Yan, Scott M Easlick and Patricia A
11. Rodrigues AD: Use of in vitro human metabolism studies in
McDonnell. Allen Reitz and Edward L Tolman are gratefully
drug development. An industrial perspective.Biochem
acknowledged for reviewing the manuscript. Pharmacol (1994) 48(12):2147-2156.
40 Current Opinion in Drug Discovery & Development 2000 Vol 3 No 1
12. Rodrigues AD: Preclinical drug metabolism in the age of
27. Bohm H-J: Computational tools for structure-based ligand high-throughput screening: An industrial perspective. design.Prog Biophys Mol Biol (1996) 66(3):197-210. Pharm Res (1997) 14(11):1504-1510.
• A good review of some of the potential problems encountered in
28. Gerhold D, Rushmore T, Caskey CT: DNA chips: promising preclinical drug metabolism and rational screening strategies.toys have become powerful tools.Trends Biochem Sci (1999) 24(5):168-173.
13. Rodrigues AD: Rational high-throughput screening in preclinical drug metabolism.Med Chem Res (1998)
29. Johnson DE, Braeckman RA, Wolfgang GH: Practical aspects 8(7/8):422-433. of assessing toxicokinetics and toxicodynamics.Curr Opin Drug Discovery Dev (1999) 2(1):49-57.
14. Tarbit MH, Berman J: High-throughput approaches for evaluating absorption, distribution, metabolism and
30. Todd MD, Ulrich RG: Emerging technologies for accelerated excretion properties of lead compounds.Curr Opin Chem toxicity evaluation of potential drug candidates.Curr Opin Biol (1998) 2:411-416. Drug Discovery Dev (1999) 2(1):58-68.
15. Smith DA, van de Waterbeemd H: Pharmacokinetics and
31. Wildman SA, Crippen GM: Prediction of physicochemical metabolism in early drug discovery.Curr Opin Chem Biol parameters by atomic contributions. J Chem Inf Comput Sci
(1999) 3:373-378.
(1999) 39(5):868-873.
16. Sinko PJ: Drug selection in early drug development:
32. Huuskonen J, Salo M, Taskinen J: Aqueous solubility Screening for acceptable pharmacokinetic properties prediction of drugs based on molecular topology and using combined in vitro and computational approaches. neural network modeling.J Chem Inf Comput Sci (1998) Curr Opin Drug Discovery Dev (1999) 2(1):42-48. 38(3):450-456.
17. Benton D: Integrated access to genomic and other
33. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ:
bioinformation: an essential ingredient of the drug Experimental and computational approaches to estimate discovery process.SAR QSAR Environmental Research solubility and permeability in drug discovery and
(1998) 8(3-4):121-155. development setting.Adv Drug Deliv Rev (1997) 23:3-25.
18. Broach JR, Thorner J: High-throughput screening for drug
34. Stenberg P, Luthman K, Ellens H, Lee CP, Smith PL, Lago A,
discovery.Nature (1996) 384:14-16.
Elliott JD, Artursson P: Prediction of the intestinal absorption of endothelin receptor antagonists using three
19. Silverman L, Campbell R, Broach JR: New assay theoretical methods of increasing complexity.Pharm Res technologies for high-throughput screening.Curr Opin
(1999) 16(10):1520-1526. Chem Biol (1998) 2:397-403.
35. Erhardt PW: Drug metabolism data: Past and present
20. Mere L, Bennett T, Coassin P, England P, Hamman B, Rink T,
status.Med Chem Res (1998) 8:400-421.
Zimmerman S, Negulescu P: Miniaturized FRET assays and microfluidics: key components for ultra-high-throughput
36. Cronin MT: Computer-aided prediction of drug toxicity in screening.Drug Disc Today (1999) 4(8):363-369. high throughput screening.Pharm Pharmacol Commun (1998) 4:157-163.
21. Winkler T, Kettling U, Koltermann A, Eigen M: Confocal fluorescence coincidence analysis: an approach to ultra
37. Kansy M, Senner F, Gubernator K: Physicochemical high high-throughput screening.Proc Natl Acad Sci USA (1999) throughput screening: Parallel artificial membrane 96(4):1375-1378. permeation assay in the description of passive absorption
• An interesting paper outlining an approach to screen 100,000processes.J Med Chem (1998) 41(7):1007-1010.
38. Plumb JA: Cell sensitivity assays: the MTT assay.Methods
22. Fecik RA, Frank KE, Gentry EJ, Menon SR, Mitscher LA,
Mol Med (1999) 8(Cytotoxic drug resistance mechanisms):25-30.
Telikepalli H: The search for orally active medications through combinatorial chemistry.Med Res Rev (1998) 18:149-185.
39. Caldwell GW, Masucci JA, Chacon E: High throughput liquid
• A good review of combinatorial chemistry with ideas concerningchromatography-mass spectrometry assessment of the the incorporation of ADME properties in the design of libraries.metabolic activity of commercially available hepatocytes from 96-well plates.Comb Chem High-Throughput Screen
23. Stanton DT, Morris TW, Roychoudhury S, Parker CN:
(1999) 2(1):39-51. Application of nearest-neighbor and cluster analyses in pharmaceutical lead discovery.J Chem Inf Comput Sci
40. Van Breemen RB, Nikolic D, Bolton JL: Metabolic screening
(1999) 39(1):21-27. using on-line ultrafiltration mass spectrometry.Drug Metab Dispos (1998) 26(2):85-90.
24. de Julian-Ortiz JV, Galvez J, Munoz-Collado C, Garcia-
• An interesting approach for doing online drug metabolism.
Domenech R, Gimeno-Cardona C: Virtual combinatorial syntheses and computational screening of new potential
41. Nikolic D, Fan PW, Bolton JL, Van Breemen RB: Screening anti-herpes compounds.J Med Chem (1999) 42(17):3308- for xenobiotic electrophilic metabolites using pulsed ultrafiltration-mass spectrometry.Comb Chem High- Throughput Screen (1999) 2(3):165-175.
25. Ekins S, Bravi G, Binkley S, Gillespie JS, Ring BJ, Wikel JH,
Wrighton SA: Three and four dimensional-quantitative
42. Crespi CL, Miller VP, Penman BW: Microtiter plate assays structure activity relationship (3D/4D-QSAR) analyses of for inhibition of human, drug-metabolizing cytochromes CYP2D6 inhibitors.Pharmacogenetics (1999) 9(4):477-489. P450.Anal Biochem (1997) 248(1):188-190.
26. Wang J, Ramnarayan K: Toward designing drug-like
43. Crespi CL: Higher-throughput screening with human libraries: A novel computational approach for prediction of cytochromes P450.Curr Opin Drug Discovery Dev (1999) drug feasibility of compounds.J Combinatorial Chem (1999) 2(1):15-19. 1(6):524-533. Compound optimization in early- and late-phase drug discovery Caldwell 41
44. Crespi CL, Miller VP, Penman BW: High throughput
56. McDonnell PA, Caldwell GW, Masucci JA: Using capillary screening for inhibition of cytochrome P450 metabolism. electrophoresis/frontal analysis to screen drugs Med Chem Res (1998) 8(7/8):457-471. interacting with human serum proteins.Electrophoresis
•• An outstanding paper for screening the inhibition of P450
(1998) 19(3):448-454.
57. Mattes WB, Li AP: Quantitative reverse transcriptase/PCR
45. Avdeef A, Box KJ, Comer JE, Gilges M, Hadley M, Hibbert C,
assay for the measurement of induction in cultured
Patterson W, Tam KY: pH-Metric log P 11. pK determination hepatocytes.Chem Biol Interact (1997) 107:47-61. of water-insoluble drugs in organic solvent-water mixtures.J Pharm Biomed Anal (1999) 20(4):631-641.
58. Sanwald-Ducray P, Dow J: Prediction of the pharmacokinetic parameters of reduced-dolasetron in man
46. Takacs-Novak K, Avdeef A: Interlaboratory study of log P using in vitro/in vivo and interspecies allometric scaling. determination by shake-flask and potentiometric methods. Xenobiotica (1997) 27(2):189-201. J Pharm Biomed Anal (1996) 14(11):1405-1413.
59. Olah TV, McLoughlin DA, Gilbert JD: The simultaneous
47. Avdeef A: pH-Metric solubility. 1. Solubility-pH profiles determination of mixtures of drug candidates by liquid from Bjerrum plots. Gibbs buffer and pK in the solid state. chromatography/atmospheric pressure chemical Pharm Pharmacol Comm (1998) 4(3):165-178. ionization mass spectrometry as an in vivo drug screening procedure.Rapid Commun Mass Spectrom (1997) 11:17-23.
48. Avdeef A, Box KJ, Comer JE, Hibbert C, Tam KY: pH-Metric logP 10. Determination of liposomal membrane-water
60. Bayliss MK, Frick LW: High-throughput pharmacokinetics: partition coefficients of ionizable drugs.Pharm Res (1998) Cassette dosing.Curr Opin Drug Discovery Dev (1999) 15(2):209-215. 2(1):20-25.
49. Yang CY, Cai SJ, Liu H, Pidgeon C: Immobilized artificial
61. Wong PS, Yoshioka K, Xie F, Kissinger PT: In vivo membranes - screens for drug-membrane interactions.Adv microdialysis/liquid chromatography/tandem mass Drug Deliv Rev (1997) 23(1-3):229-256. spectrometry for the on-line monitoring of melatonin in rat. Rapid Commun Mass Spectrom (1999) 13(5):407-411.
50. Caldwell GW, Masucci JA, Evangelisto M, White R: Evaluation of the immobilized artificial membrane
62. Masucci JA, Ortegon ME, Jones WJ, Shank RP, Caldwell GW:
phosphatidylcholine. Drug discovery column for high- In vivo microdialysis and liquid chromatography/ performance liquid chromatographic screening of drug- thermospray mass spectrometry of the novel membrane interactions.J Chromatogr A (1998) 800(2):161-169. anticonvulsant 2,3:4,5-bis-O-(1-methylethylidene)-β-D- fructopyranose sulfamate (topiramate) in rat brain fluid.J
51. Ducarne A, Neuwels M, Goldstein S, Massingham R: IAM Mass Spectrom (1998) 33(1):85-88. retention and blood brain barrier penetration.Eur J Med Chem (1998) 33:215-223.
63. Obach RS, Baxter JG, Liston TE, Silber BM, Jones BC,
MacIntyre F, Rance DJ, Wastall P: The prediction of human
52. Masucci JA, Caldwell GW, Foley JP: Comparison of the pharmacokinetic parameters from preclinical and in vitro retention behavior of β-blockers using immobilized metabolism data.J Pharmacol Exp Ther (1997) 283(1):46-58. artificial membrane chromatography and lysophospholipid micellar electrokinetic chromatography.J Chromatogr A
64. Iwatsubo T, Hirota N, Ooie T, Suzuki H, Shimada N, Chiba K,
(1998) 810(1-2):95-103.
Ishizaki T, Green CE, Tyson CA, Sugiyama Y: Prediction of in vivo drug metabolism in the human liver from in vitro
53. Yee S: In vitro permeability across Caco-2 cells (colonic) metabolism data.Pharmacol Ther (1997) 73(2):147-171. can predict in vivo (small intestinal) absorption in man - fact or myth.Pharm Res (1997) 14(6):763-766.
65. Houston JB, Carlile DJ: Prediction of hepatic clearance from microsomes, hepatocytes, and liver slices.Drug Metab Rev
54. Irvine JD, Takahashi L, Lockhart K, Cheong J, Tolan JW,
(1997) 29(4):891-922.
Selick HE, Grove JR: MDCK (Madin-Darby canine kidney) cells: A tool for membrane permeability screening.J
66. Meier J: Pharmacokinetic comparison of pindolol with Pharm Sci (1999) 88(1):28-33. other beta-adrenoceptor-blocking agents.Am Heart J (1982) 104(2):364-373.
55. Caldwell GW, Easlick SM, Gunnet J, Masucci JA, Demarest K:
In vitro permeability of eight β-blockers through Caco-2
67. Hinderling PH, Schmidlin O, Seydel JK: Quantitative monolayers utilizing liquid chromatography/electrospray relationships between structure and pharmacokinetics of ionization mass spectrometry.J Mass Spectrom (1998) beta-adrenoceptor blocking agents in man. J 33(7):607-614. Pharmacokinet Biopharm (1984) 12(3):263-287.
BSR guidelines for prescribing TNF- a blockersin adults with ankylosing spondylitis. Report of aworking party of the British Societyfor RheumatologyA. Keat, N. Barkham1, A. Bhalla2, K. Gaffney3, H. Marzo-Ortega1,S. Paul4, F. Rogers5, M. Somerville3, R. Sturrock6 and P. Wordsworth7on behalf of the BSR Standards, Guidelines and Audit Working GroupTwo TNF-blocking drugs are now licensed for the
Acesso a medicamentos: Indonésia abre importante precedente a ser seguido, mas o Brasil caminha em outra direção. Começa a vir a público uma medida adotada na Indonésia, o quarto país mais populoso do planeta, para viabilizar a ampliação do acesso ao tratamento para Aids e Hepatite B no país. Em setembro, o presidente Dr. H. Susilo Bambang Yudhoyono assinou um decreto auto
 Compound optimization in early- and late-phase drug discovery: Acceptable
Compound optimization in early- and late-phase drug discovery: Acceptable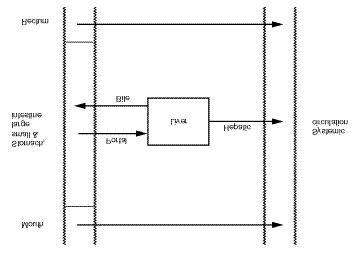 32 Current Opinion in Drug Discovery & Development 2000 Vol 3 No 1
32 Current Opinion in Drug Discovery & Development 2000 Vol 3 No 1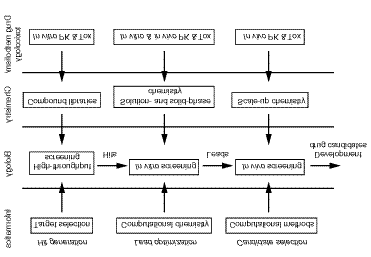 34 Current Opinion in Drug Discovery & Development 2000 Vol 3 No 1
34 Current Opinion in Drug Discovery & Development 2000 Vol 3 No 1